
Abstract
JC virus-specific CD8+ cytotoxic T lymphocytes are associated with a favorable outcome in patients with progressive multifocal leukoencephalopathy. However, very few JC virus T cell epitopes restricted to MHC class I have been defined. Of the two HLA-A*0201-restricted JCV epitopes, VP1p36 and VP1p100, studies have shown that they are conserved T cell epitopes of polyomaviruses. The cross-recognition associated to these epitopes has complicated the efforts of understanding the dynamics of immune response to JC virus. Based on the previously identified HLA-A*0201 binding T cell epitope of Simian virus 40 T antigen P281–289 (KCDDVLLLL) and BK virus T antigen P558–566 (SLQNSEFLL), T cell epitopes of JC Virus T antigen P282–290 (KCEDVFLLM) and P557–565 (SLSCSEYLL) were identified. In this report, we demonstrated that JC Virus P282–290 and P557–565 were able to stimulate T cell responses in healthy donors’ PBMCs and CD8+ cytotoxic T lymphocytes raised with both peptides could recognize and lyse their targets. Most importantly, there were no T cell cross-recognitions between JC Virus, BK Virus and SV40 virus. Therefore, JCV T-ag epitopes P282–290 and P557–565 could be better antigen epitopes compared to VP1p36 and VP1p100 to study the dynamics of cellular immune response to JCV in PML patients. In addition, as a HLA-A*0201 binding T cell epitope, both peptides could be a valuable component of immunotherapies aiming at increasing the cellular immune response against JCV for the treatment of progressive multifocal leukoencephalopathy.
Introduction
JC virus (JCV), a small ubiquitous DNA virus, causes primary asymptomatic infection during late childhood. In most individuals, the virus is quiescent in the kidney or lymphoid organs. In immunocompromised patients, JCV reactivation can cause progressive multifocal leukoencephalopathy (PML), particularly among HIV-infected individuals. It has also recently been reported in patients with multiple sclerosis or Crohn's disease treated with natalizumab (Berger and Koralnik, 2005; Safak and Khalili, 2003). The incidence of PML is 5.1% in patients with AIDS (Power et al. 2000) and 3.3% in patients with hematological malignancies (Garcia-Suarez et al. 2005). Currently there are no specific therapies for PML. Of the treatment approaches attempted for PML, including cytarabine, alfa-interferon or cidofovir, none has been convincingly shown to confer any benefit on patients’ survival (Gasnault et al. 2001; Marra et al. 2002; Geschwind et al. 2001). On the other hand, cohort studies have shown that Highly Active Antiretroviral Therapy (HAART) confers an improved survival in about 50% of HIV-infected patients (De Luca et al. 2000; Clifford et al. 1999; Tassie et al. 1999). The survival benefit conferred by HAART is largely attributed to the immune reconstitution induced by antiretroviral therapy. This is consistent with the findings from previous studies (Koralnik et al. 2002; Du Pasquier et al. 2004) that the detection of peripheral blood CD8+ cytotoxic T lymphocytes (CTLs) against JCV VP1 epitopes VP1p36 and VP1p100 is associated with a better outcome in PML patients. Therefore prospective follow-up of the frequency and phenotype of JCV-specific CD8+ T-cell response in these patient populations at risk would not only provide valuable information on the dynamic interaction between the virus and the host, but also provide a useful prognostic tool in the clinical management of these patients.
Previous studies (Krymskaya et al. 2005; Chen et al. 2006) showing that VP1p36 and VP1p100 are conserved T cell epitopes of BK virus (BKV) and JCV suggested that the immune response against JCV may be mediated by CTLs specific for BKV and vice versa. Unfortunately, this cross-immunity originated from 75% sequence homology between BKV and JCV has made it a difficult task to gain a better understanding of the dynamics of cellular immune response to JCV in patients with PML. To address these challenges, we sought to study the T cell response to JCV T antigen (T-ag), particularly the non-conserved T cell epitopes of BKV and JCV. Here we present our findings of two non-conserved HLA-A*0201 binding T cell eptitope of JCV T-ag. These two non-conserved T cell eptitopes of JCV T-ag are derived from the previous report of HLA-A*0201 binding T cell epitope BKV P558–566 (SLQNSEFLL) (Provenzano et al. 2006) and Simian virus 40 (SV40) (KCDDVLLLL) (Schell et al. 2001) since human polyomaviruses shares approximately 70% sequence homology with SV40 (Butel and Lednicky, 1999). A non-conserved JCV T cell epiotpe could serve a better marker to study human cellular immune response to JCV and be a valuable tool for immunotherapies aiming at increasing the cellular immune response against JCV in the treatment of HIV + individuals with PML.
Materials and Methods
Study Population
Peripheral blood mononuclear cells (PBMCs) were harvested by leukapheresis from paid healthy donors at Thomas Jefferson University and National Institute of Health (NIH). Lymphocytes and monocytes that were seperated by Elutra® (Gambro) were processed by Ficoll-Hypaque, frozen and stored in liquid nitrogen until use.
Peptides, Cell Lines and Viral Lysates
The T antigen sequences of JCV, BKV and SV40 are compared directly and the JCV T cell epitope sequences are derived from the similar locations. Further, the putative HLA-A*0201 binding motifs (9-mer peptides) were examined using the SYFPEITHI epitope-prediction program (http://www.syfpeithi.de). The corresponding sequences of previously identified SV40 (P281–289) epitope KCDDVLLLL (Schell et al. 2001) in BKV and JCV T-ag are KCEDVFLLL (BKV P283–291) and KCEDVFLLM (JCV P282–290), respectively. The corresponding sequences of previously identified BKV (P558–566) epitope SLQNSEFLL (Provenzano et al. 2006) in JCV and SV40 are SLSCSEYLL (JCV P557–565) and CLERSEFLL (SV40 P556–564), respectively. These peptides along with Cytomegalovirus (CMV) pp65 HLA-A*0201 binding peptide P495–503 NLVPMVATV were purchased from Biosynthesis Inc (>90% purity). The T2 (HLA-A2+) transport-deletion mutant B-T cell hybrid cell line and the C1R-A2 cell line, which is a human B-cell lymphoblastoid line transfected to express surface HLA-A2 antigen were gifts from Dr. John Barrett, National Heart, Lung, and Blood Institute, NIH. EBV-transformed B cell lines (LCL) was generated as described before (Li et al. 2007). Briefly, 5 to 10 × 106 PBMC was incubated with concentrated supernatant of B95–8 cultures, in the presence of 1μg/mL cyclosporin A (Sandoz) to establish an LCL. JCV lysates were prepared by freeze—thaw lysis of a JCV-infected embryonic kidney cell line, a gift from Dr. Eugene Major, National Institute of Neurological Disorders, NIH.
Preparation of Dendritic Cells (DCs) Expressing Human Polyomavirus Antigens
DCs were generated from PBMCs as described previously (Li et al. 2006). Briefly, 10 × 106 elutriated monocytes were thawed and added immediately to one well of a tissue-culture-treated six-well plate (Falcon, Becton Dickinson) in 3 ml culture medium consisting of RPMI 1640 medium (Mediatech, Inc) containing 10% heat-inactivated normal AB serum (Gemini) and DNase I (960 U) (Worthington Biochemical Corporation). After 90 min incubation at 37 °C, non-adherent cells were discarded. The adherent cells were washed three times with warm phosphate-buffered saline (PBS, Mediatech, Inc). Afterwards, human recombinant interleukin-4 (rIL-4; BD PharMingen) at a final concentration of 500U/ml and human recombinant granulocyte—monocyte colony-stimulating factor (GM-CSF; Sandoz Pharmaceuticals) at a final concentration of 800 U/ml were added. After 5 days incubation, immature DCs were pulsed with JCV lysate. After another 4-5 days culture, they were ready as DCs expressing human polyomavirus antigens.
Generation of Polyclonal Polyomavirus-Specific Cytotoxic T Cells
PBMCs were co-cultured with autologous monocytes that were previously pulsed with peptide (2 μM) at a responder/stimulator ratio of 10:1. Starting on day 7, the responder cells were re-stimulated with monocytes pulsed with peptide at a responder/stimulator ratio 10:1. Also, on day 7 of culture and every 3 days thereafter, recombinant human IL-2 (rhIL-2; Proleukin; Chiron), at a final concentration of 20 U/ml, was added to expand the proliferating T cells. On day 14, the frequency of antigen-specific T cells was determined by flow cytometry. If the frequency of peptide specific T cells was not adequate, lymphocytes were stimulated again with monocytes pulsed with peptide at a responder/stimulator ratio 10:1. Cells were cultured in the presence of IL2 for another week before they were used in the experiments.
HLA-A*0201 Restriction of JCV P282–290 and P557–565
Peptide-loaded transformed lymphoid cell lines were used as antigen presenting cells (APCs) to define the HLA restriction. Positive controls were peptideloaded T2 and C1R-A2 sharing HLA-A02 allele with the responder and negative control was peptideloaded LCL (HLA-A02-) sharing no HLA alleles with the responder. These APCs were pulsed with 5 μM of the peptide of interest or with an irrelevant peptide CMV pp65 P495–503 for 2 hours. After being washed at least twice with warm culture media, 10 6 APCs were used to stimulate equal numbers of responder lymphocytes. IFN-γ production was measured by flow cytometry.
Intracellular Cytokine Staining (ICC)
Intracellular IFN-γ staining was performed as described (Li et al. 2006). Briefly, 2 × 106 lymphocytes were cultured with irradiated (50 Gy) peptide-pulsed-T2 cell lines with a ratio of 1/1. After 2 hours, 10 μg/ml brefeldin A (Sigma) was added and cultures were incubated for a further 4 hours. After this period, cells were stained with CD3-PerCP and CD8-PE, fixed in FACS Lyse, permeabilized with FACS Perm2 and labeled with anti-IFN-γ-fluorescein isothiocyanate (all from Becton Dickinson) according to the intracellular-staining procedure protocol supplied by the manufacturer. Immunofluorence measurements were acquired on a FACScalibur flow cytometer and analyzed using CellQuest software. The frequency determined for the control (T2 cell line loaded with CMV pp65 P495–503) was subtracted from the frequency for the target peptide-stimulated population to determine the true polyomavirus antigen-specific T-cell frequency.
Chromium Release Assay
The cytotoxic activity of T cells was measured by a 4 hours 51chrom (Cr) release assay as described previously (Brunner et al. 1968) using effector/target ratios of 30:1, 10:1 and 3:1. The following target cells were tested: T2 cell line pulsed with polyomavirus target peptides (2μM): BKV P283–291, JCV P282–290, SV40 P281–289 and BKV P558–566, JCV P557–565, SV40 P556–564.
Results
T Cell Responses to JCV P282–290 and JCV P557–565
PBMCs from 6 healthy donors (HLA-A*0201+) were used to study T cell responses to JCV P282–290 and P557–565. Each experiment was performed at least 3 times. First, PBMCs were screened directly by flow cytometry to detect T-cell responses to these peptides. No significant IFN-γproduction was identified in these individuals (data not shown). In subsequent experiments, monocytes were pulsed with peptide to stimulate autologous lymphocytes initially. On day 7, lymphocytes were re-stimulated with peptide-loaded monocytes, followed by expansion in the presence of IL2. On day 14, significant IFN-γ production from CD3+CD8+ T cells (usually < 1%) was identified in 3 healthy donors when these lymphocytes were exposed to JCV P282–290 and P557–565 (Table 1). After 4 weekly stimulation and expansion, the frequency of JCV P282–290 and P557-565-specific T cells could be increased to 25% (Figs. 1 and 2).
Frequency of JCV-specific T cells after 2 weekly stimulations.
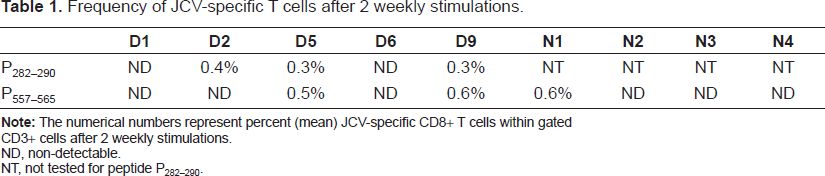
CD3+ cells after 2 weekly stimulations.
ND, non-detectable.
NT, not tested for peptide P282–290.
Identification of HLA Class I Restriction of Peptide JCV P282–290 and P557–565
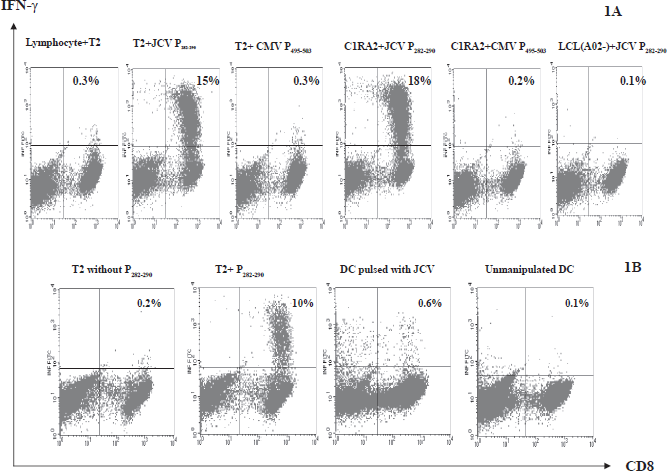
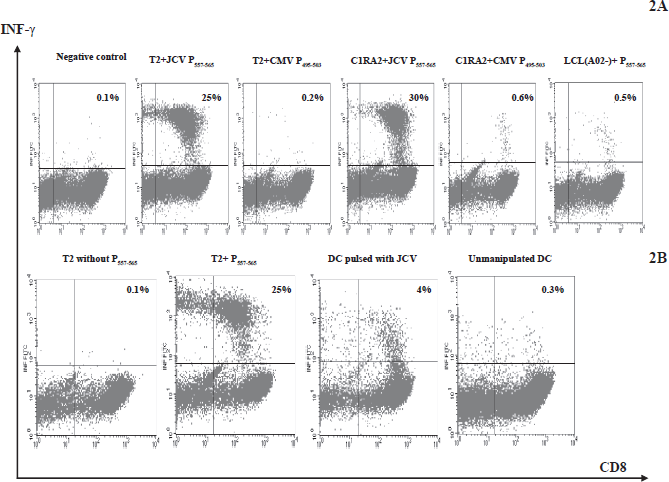
A panel of transformed lymphoid APCs matched (T2 and C1R-A2) or mismatched (donor 7 LCL, HLA-A02-) with the responder for HLA-A02 alleles were used to define the MHC restriction of JCV P282–290 and JCV P557–565. Lymphocytes from HLA-A*0201 individuals showed significant IFN-γ production with JCV P282–290 and JCV P557–565, but not with an irrelevant peptide CMV pp65 P495–503 (Figs. 1A and 2A).
JCV P282–290 and P557–565 is Naturally Expressed in APCs
T cells from healthy donor 5 (HLA-A*0201+) were raised with JCV P282–290. After 4 weekly stimulations with peptide-loaded-monocytes, the frequency of JCV P282–290-specific T cells was increased to 10% (Fig. 1B). Autologous immature DCs were prepared as described in Methods Section. After immature DCs were pulsed with JCV and cultured for 5 days, they were co-cultured with CTLs raised with JCV P282–290 for intracellular cytokine staining. Representatives of the intracellular cytokine staining results are shown in Figure 1B.
Similarly, T cells from healthy donor 5 (HLA-A*0201+) were raised with JCV P557–565. After 4 weekly stimulations with peptide-loaded-monocytes, the frequency of JCV P557–565-specific T cells was increased to 25% (Fig. 2B). After immature DCs were pulsed with JCV and cultured for 5 days, they were co-cultured with CTLs raised with JCV P557–565 for intracellular cytokine staining. Representatives of the intracellular cytokine staining results are shown in Figure 2B.
JCV P282–290 and P557–565 are Non-Conserved T Cell Epitopes Derived from T-ag
T cells from healthy donor 5 were raised with JCV P282–290 for 4 weeks. T-cell cross-recognition experiments by intracellular IFN-γ staining of the three T-ag epitope peptides were performed three times. T2 cell lines pulsed with BKV P283–291, JCV P282–290 and SV40 P281–289 were used as APCs. There was no significant IFN-γproduction when T cells raised with JCV P282–290 were exposed to T2 cell lines loaded with BKV P283–291 and SV40 P281–289. Representatives of cross recognition experiment results are shown in Figure 3A. Cytotoxicity studies were shown in Figure 3B. JCV P282–290-specific CTLs lysed T2 pulsed with JCV P282–290 preferentially over T2 pulsed with BKV P283–291 and SV40 P281–289. Unfortunately, BKV P283–291-specific T cells could not be generated in PBMCs by repeated stimulations with monocytes loaded with BKV peptide.
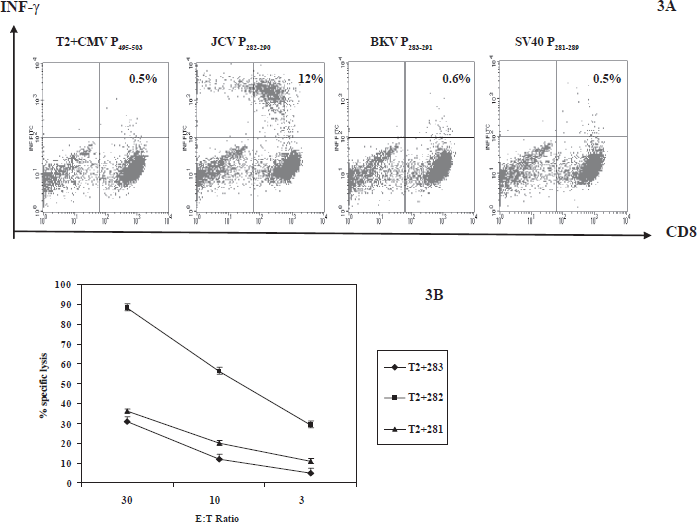
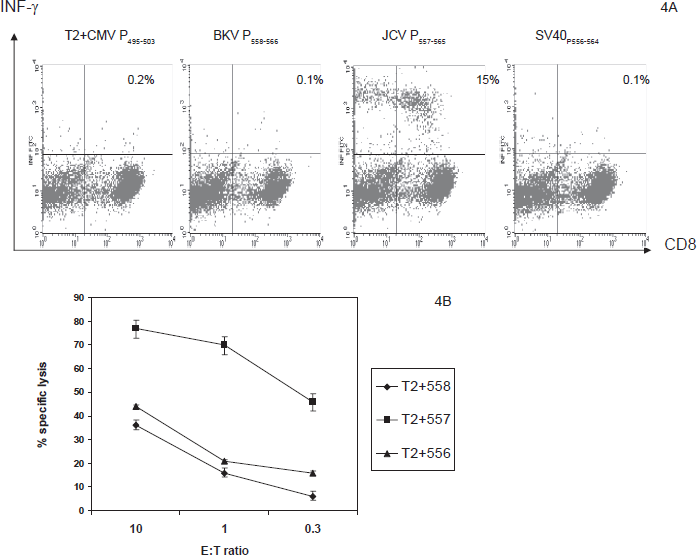
Similarly, T cells from healthy donor 5 were raised with JCV P557–565 for 4 weeks. T2 cell lines pulsed with BKV P558–566, JCV P557–565 and SV40 P556–564 were used as APCs. There was no significant IFN-γ production when T cells raised with JCV P557–565 were exposed to T2 cell lines loaded with BKV P558–566 and SV40 P556–564. Representatives of cross recognition experiment results are shown in Figure 4A. Cytotoxicity studies were shown in Figure 4B. JCV P557–565-specific CTLs lysed T2 pulsed with JCV P557–565 preferentially over T2 pulsed with BKV P558–566 and SV40 P556–564. BKV P558–566-specific T cells could also be raised in PBMCs by repeated stimulations with monocytes loaded with BKV peptide P558–566. These T cells could recognize T2 cell lines loaded with P558–566 but not with JCV P557–565 or SV40 P556–564 (data not shown). In addition, these T cells also preferentially lysed T2 cell lines loaded with BKV P558–566 over T2 pulsed with JCV P557–565 or SV40 P556–564 (data not shown).
Discussion
Previous studies (Schell et al. 2001; Provenzano et al. 2006) have identified HLA-A*0201 binding T cell epitope of SV40 P281–289 and BKV P558–566 from T-ag. Based on these findings, we identified the corresponding sequences of JCV P282–290 and P557–565 and tested their ability to induce T cell response in healthy donors. No JCV peptide specific T cells could be detected by flow cytometry in unmanipulated PBMCs from 6 healthy donors. However, it was possible to detect JCV P282–290 and P557–565-specific T cells after stimulating T cells with JCV peptides for 2 weeks (usually less than 1%). These results are consistent with the findings in our previous studies (Li et al. 2006). This low magnitude of the CD8+ T-cell response against JCV could be attributed to viremia that rarely occurred in immunocompetent individuals (Koralnik et al. 1999) following the primary infection that usually happened during childhood (Demeter, 2000). Therefore a small virus-specific memory cell population may be sufficient to prevent widespread viral replication in healthy individuals. As JCV antigen-specific T cells were elicited directly with peptides using PBMCs as nonprofessional APCs, we presume that the T-cell responses described here were derived from circulating memory T cells rather than from naive T cells, but the low frequencies in the blood precluded study of their memory-effector phenotype. The fact that CTLs raised with synthetic peptides recognized dendritic cells, which were pulsed with JCV lysates suggests that these epitopes of human polyomaviruses were processed and presented naturally by APCs. Of note, we do not believe that the JCV lysate contains a significant amount of T-ag protein or fragments thereof to be processed and presented by dendritic cells directly, because there was no T cell recognition of dendritic cells pulsed with JCV until day 4–5 after viral lysate was added to immature dendritic cells (unpublished data). In addition, the quantity of IFN-γ producing cells were tripled on day 5 compared that on day 4 when CTLs were co-cultured with immature DCs pulsed with JC virus (unpublished data).
Because the genomes of human polyomaviruses and SV40 are highly similar, previous studies (Krymskaya L et al. 2005; Chen et al. 2006; Li et al. 2006) have shown that T cell cross-recognition of VP1 and T-ag epitopes exists between BKV, JCV and SV40. T cell epitope cross-recognition has made it a difficult task to gain a better understanding of the dynamics of cellular immune response to JCV in patients with PML. In one report using conserved T cell epitopes JCV VP1p36 and VP1p100 to study the frequency and phenotype of JCV-specific CD8+ CTLs in the peripheral blood of patients with PML, BKV load has to be measured in order to exclude compounding cross immune response (Lima et al. 2007). Therefore it is much desirable to study the dynamics of cellular immune response to JCV with non-conserved T cell epitopes. In this report we sought to identify these non-conserved T cell epitopes of T-ag instead of VP1. T-ag is expressed early in virus infection and regulates viral transcription by binding the regulatory region of the genome. As the concentration of large T-ag builds up in the nucleus, transcription of the early genes is switched to DNA replication. Subsequently, transcription of the late genes occurs from the late promoter, resulting in the synthesis of the structural proteins VP1, VP2 and VP3. Therefore CTLs specific to T-ag should be activated early than that of CTLs specific to VP1 and before virus particles were packaged and released from the host cells. In theory CTLs specific to T-ag could provide better cellular immune protection and clearance of viremia in the host. Nevertheless, our findings here could truly represent an advance to study and follow immune response to JCV with the non-conserved epitope compared to conserved epitope in patients with PML. In addition, it could also be a valuable tool for designing immunotherapies aiming at increasing the cellular immune response against JCV in the treatment of HIV + individuals with PML (Yang et al. 2007). Meanwhile, we also would like to point out the weakness of our study here. That is our data were generated mainly from healthy donors without being confirmed in patients with active PML and HIV infection. This becomes particularly important in view that changing immunodominance patterns in antiviral CD8+ T-cell responses after loss of epitope presentation or chronic antigenic stimulation has been reported in murine models (van der Most et al. 2003).
Recently, a few studies have implicated JCV in the human carcinogenesis, particularly of GI tract tumors (Ricciardiello et al. 2001; Enam et al. 2002; Casini et al. 2005; Shin et al. 2006). The identification of non-conserved JCV T-ag epitope here provides a potential opportunity to examine the presence of JCV T-ag in tumor tissues with T cell recognition approach. The discovery that apoptotic material could be processed and cross-presented by DCs to stimulate specific HLA-restricted CD8 + T cells (Albert et al. 1998) thus makes it possible to use antigen-specific T cells to examine the presence of JCV T-ag in the tumor cells. In addition, by using DCs to process the apoptotic tumor tissues or lysates and present to autologous CTLs, the MHC restriction associated with HLA phenotype of tumor tissue would become irrelevant. Indeed, CTLs raised with BKV T-ag peptide LPLMRKAYL, which is a conserved T cell epitope for JCV and SV40 (Li et al. 2006), are able to recognize autologous DCs loaded with apoptotic COS-1 cells and/or their lysates (unpublished data). Demonstration of T-ag expression in tumors using the exquisite sensitive and specific T cell recognition could have profound impact on our thinking about viral carcinogenesis and therapy. Recently we developed a novel solid phase T cell selection method so that large scale highly purified tumor antigen-specific T cells can be prepared within 2–3 weeks (Li et al. 2007; Li et al. 2008). Large scale highly purified BKV or JCV-specific T cells can also be generated in 2–3 weeks by using this new T cell purification method (Li et al. 2006). Adoptive immunotherapy for GI tract tumors implicated by JCV could potentially have profound clinical impact since there is no cure for these cancers with advanced stage.
In summary, we have identified non-conserved HLA-A*0201 binding T cell epitopes of JCV T-ag P282–290 and P557–565. These peptides are better antigen epitopes than VP1p36 and VP1p100 to study the dynamics of cellular immune response to JCV in PML patients. Because of its nature as a non-conserved T cell epitope, it eliminates compounding cross immune contribution from BKV and can significantly simply the study process. In addition, as a HLA-A*0201 binding T cell epitope, it can be a critical component of immunotherapies aiming at increasing the cellular immune response against JCV for the treatment of PML.
Financial Support
Department of Health, Pennsylvania.
Financial Disclosure
All authors have declared there are no financial conflicts of interest related to this work.
The authors have no commercial or other association that might pose a conflict of interest.
Footnotes
Acknowledgement
We are grateful to Dr. Takami Sato, Department of medical Oncology, Thomas Jefferson University Hospital for the statistical analysis.