
Abstract

Kaposi's sarcoma (KS) was originally described in 1872 by the Hungarian dermatologist Moritz Kaposi as an idiopathic, multiple pigmented sarcoma of the skin. This type of KS, later classified as classic KS, predominantly occurred in elderly people, particularly men of Mediterranean, Eastern European, or Jewish descent. A high occurrence of African or endemic KS was reported during the early 1950's in equatorial African countries, while during the 1960's KS emerged among immune-suppressed patients following organ transplantation. A tremendous increase in the incidence of KS was noticed during the early 1980's in parallel with the acquired immunodeficiency syndrome (AIDS) epidemic, leading to a formal recognition of KS as an AIDS-associated malignancy. The geographic variations in the incidence of KS together with the relatively high occurrence of AIDS-KS, particularly among homosexual men, suggested the existence of a unique KS infectious agent (7, 8). In 1994 Chang and colleagues, amplified two novel DNA sequences from an AIDS-associated KS lesion, by using a subtractive hybridization technique (representational difference analysis (RDA)). These sequences displayed homology to herpesviral capsid and tegument genes and led to the complete sequencing of the 165 Kbp viral genome of the newly discovered virus, Kaposi's Sarcoma-Associated Herpesvirus (KSHV) or human herpesvirus 8 (HHV-8). KSHV was established as being present in virtually all pathologically confirmed KS specimens (11, 12).
Treatment with highly active antiretroviral therapy (HAART) has been associated with a dramatic decline in the incidence of KS (31), so that AIDS-associated KS is uncommon overall in the Western world. Sub-Saharan Africa has the highest rates of infection (due mostly to the high rates of AIDS) with many countries experiencing sera-prevalence rates exceeding 60%, so that in certain African countries it is now the most common cancer in adult men, and the second most common in adult women (44). The incidence of KS among transplant recipients is proportional to the ethno-geographical prevalence of KSHV, ranging from 0.5% in most Western countries to 5.3% in Saudi Arabia (50). In addition to KS, which is a tumour of endothelial cell origin, several haematological predominantly B cell disorders are also associated with KSHV. The two most well characterized are AIDS-associated primary effusion lymphomas (PEL) and a subset of multicentric Castleman's disease (MCD), an aggressive lymphoproliferative disorder.
PELs are monoclonal, non-Hodgkin's B cell lymphomas that are commonly co-infected with EBV. Cell lines established from PEL, unlike KS tumour explants, stably maintain viral episomes at high copy number (30–150 copies per cell) and are the source of virus for most virologic and serologic studies. Most studies on latency of gammaherpesviruses (EBV) used lymphoblastoid cell lines that were derived from lymphomas or were established by in vitro infection/immortalization of peripheral blood lymphocytes. In vitro immortalized cells lines can be cultured indefinitely. PEL cell lines that carry latent KSHV have been successfully established, although no in vitro immortalization of B lymphocytes has been reported yet (10, 44).
KSHV viral genome consists of a ~140kb long unique coding region flanked by a multiple GC rich ~800bp TRs (terminal repeats) giving a total estimated size of around 170kb (57, 60). In analogy with the biology of EBV, it was shown early on after the discovery of KSHV DNA by Chang et al. (11) that the restricted gene expression of KSHV in KS-lesions exhibited properties of a latent infection (73). In situ hybridization and RT-PCR revealed that the majority of cells in KS lesions (10), and in PEL-derived cell lines are latently infected (58), although only around 10% of spindle and endothelial cells are KSHV positive (20, 30). In late stage nodular lesions around 90% of spindle cells contain KSHV suggesting that the virus provides a growth advantage to infected cells (10, 57, 66, 67). Primary infection by KSHV leads to the persistence of the viral genome as a circular DNA episome in the host cell.
The majority of latently infected cells express only a few viral genes, which play an important role in maintaining and replicating the latent viral episome, as well as evading the immune response. The antigen was named latency-associated nuclear antigen (LANA1) (23, 34), and is encoded by open reading frame 73 (ORF73) of KSHV (33, 35, 55). The ORF73 gene is located in a cluster of four latency-associated genes and is expressed from a bi-cistronic singly spliced mRNA of 5.7kb that also encodes ORF72, a viral cyclinD homologue, ORF71, a v-FLIP protein and the kaposin gene (12, 18, 73). LANA1 is a multifunctional protein involved in viral episomal replication and maintenance by tethering the genome to host chromosomes in addition to its function as a transcription regulator (4, 68). LANA1 transforms cells by targeting p53 and preventing its induced cell death (22). In addition, LANA1 binds to the retinoblastoma protein (pRb), enabling the transcription of genes needed for cell cycle progression (54). LANA2 encoded by ORF K10.5 is expressed latently in B cells but not in KS cells. This protein inhibits p53 mediated trans-activation and apoptosis (59). vCyc encoded by ORF72 is a viral homologue of the human Cyclin D, able of mimicking its function by binding to cyclin dependent kinase 6 (Cdk6) and inactivating pRb, causing cell cycle progression (46). ORF K13, also termed ORF71, encodes the anti-apoptotic Fas-ligand IL-1 β-converting enzyme inhibitory protein (vFLIP), a homologue of the cellular protein FLIP. vFLIP is thought to protect cells from Fas and tumour necrosis factor receptor (TNFR1)-mediated apoptosis and activate NF-κB, enabling the establishment of tumours (19). LAMP is a membrane protein with similarity to LMP1 and LMP2A proteins of EBV (25). ORF K12 encodes for Kaposin A, expressed in most KSHV infected cells (36, 37, 38). These are just some examples of work with proteins which have a role in the interaction and replication of KSHV.
Replication of the KSHV Genome
Long-term maintenance of viral DNA in latently infected cells can be divided into two steps. First, viral DNA needs to be replicated by the cellular replication machinery and in second, viral genomes have to be faithfully segregated during mitosis. Ballestas et al. showed that plasmids containing two copies of terminal repeats (TR) sustain LANA1 dependent long term maintenance in BJAB (human lymphoid) cells (4). Several laboratories prepared TR containing plasmids and tested their replication in the presence and absence of LANA1 in several cell lines (13, 27, 29, 39). These experiments showed that replication of plasmids containing two copies of TR is strictly dependent of the presence of LANA1, indicating that LANA1 mediates episomal replication through DNA sequences that are located within the TR. Later on, deletion analysis showed that a 105bp TR sub-fragment replicates with identical activity of the full TR when in the presence of LANA1 (29).
LANA1 can be divided into three domains (see fig. 1): a proline and serine rich N terminal region (312aa); a variable central region (differs between isolates), that is composed of several acidic repeats including a leucine zipper motif (allows oligomerization of DNA binding proteins); and a conserved C-terminal domain, containing both a proline-rich region and a region rich in charged and hydrophobic amino acids.
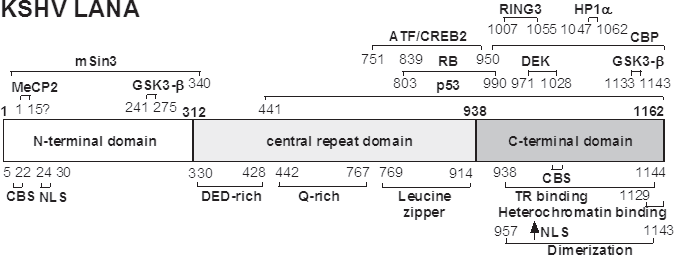
Functional protein domains of LANA1. Aminoacid residues are indicated for the boundaries of protein domains for DNA binding, dimerization, chromosome binding sites (CBS), nuclear localization signals (NLS), or protein-protein interactions (as indicated). LANA aminoacid sequence is based on sequence published by Russo et al. (Russo, 1996). Figure adapted from Lieberman et al.
LANA1 and the Cell Cycle
LANA1 is able to modulate several cellular pathways involved in cell cycle regulation and progression, particularly those involving the tumour suppressor's p53 and pRb proteins, just like other gammaherpesvirus proteins (9, 40, 48, 49). It interacts with the pRb (54) and the p53 protein (22, 32), through the region containing the C-terminus of LANA's leucine zipper of LANA. These are crucial targets for DNA virus oncogenic proteins as they are responsible for important functions in the regulation of cell cycle (42, 43).
Activation of p53 results on transcription of the p21CIP cdk inhibitor that binds to and inhibits cyclin-dependent kinases (cdks) resulting in the hypophosphorylation of pRb. When unphosphorylated, pRb interacts with E2F transcription factors preventing their release and therefore, blocking the G1-S cell cycle transition (3, 61). When phosphorylated by cdks, pRb no longer binds to E2F and the cell cycle proceeds through G1-S checkpoint (see Fig. 2).
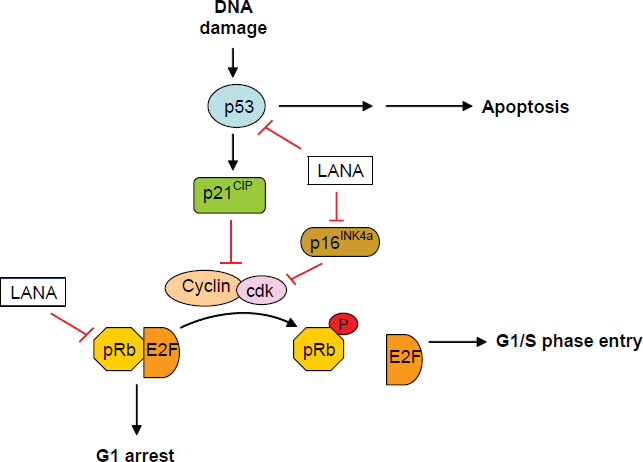
Role of LANA1 in the p53 and pRb-E2F pathways. LANA1 interacts with p53 therefore blocking p53-mediated apoptosis and G1 cell cycle arrest. Interaction with pRb releases E2F and leads to S phase entry. LANA1 interference with p16INK4a also conduces to cell cycle progression.
The interaction of LANA1 with p53 represses its transcriptional activity and its ability to induce cell death, but does not influence p53 DNA-binding ability nor induces its degradation (22). This was also shown for EBV LMP1 protein, except that repression is not through direct interaction but through the NF-κB pathway (40). The interaction of LANA1 with pRb leads to the up-regulation of elongation factor 2 (E2F)-dependent transcription, activating genes that are required for progression into S phase of the cell cycle (56, 54). EBV EBNA3C can also interact with pRb and provide resistance to p16INK4a over-expression in fibroblasts (49). In addition, LANA1 expression was shown to protect lymphoid cells from p16INK4a -induced cell cycle arrest and induce S-phase entry (2). The targeting of this cdk inhibitor is also common to EBV (47, 52). LMP1 is able to suppress the senescence-associated induction of p16INK4a and its expression in fibroblasts (69, 71), occurring in part by blocking the downstream mediator of the p16INK4a -RB pathway, that belongs to the E2F family of transcription factors (45) and therefore providing the virus with another mean to induce carcinogenesis. More recently, it was demonstrated that constitutive expression of LANA1, particularly its C-terminus domain, induces chromosomal instability through inhibition of p53 function generating an increase in cell proliferation and entry into S phase (64). Consistent with the interference with the p53 and pRb pathways, LANA1 has been shown to induce primary rat embryo fibroblast cells transformation (54), corroborating its oncogenic potential. In agreement with p53 inhibition, Curreli and colleagues demonstrated that down-regulation of LANA1 expression in KSHV-infected B cells by glicyrihizic acid restores p53 transcriptional function and consequently induces cell cycle arrest at the G1 checkpoint and triggers apoptotic signals (16).
The inactivation of both the p53 and pRb tumour suppressors and its pathways are common to DNA oncogenic viruses for cell proliferation induction and cancer development. Thus, by altering regulation of these cell cycle checkpoints and apoptotic regulators, LANA1 promotes cell survival, contributing to cell proliferation and to viral persistence during latency. For that reason, inhibition of these LANA1 functions would be important to overcome KSHV-associated cancers (74, 75).
LANA1 Inhibition
LANA1 displays a punctuate staining pattern in the nucleus of KSHV infected cells in an immunofluorescence antibody assay (IFA) (23, 33, 35, 55). Such patterning is due to the co-localization of LANA1 with KSHV episomes on cellular chromosomes, implicating LANA1s involvement in the replication and maintenance of KSHV episomes in latently infected cells (4). In PEL-derived cell lines, some 30 to 150 copies of the episome are maintained in the nucleus of each infected cell as circular mini-chromosomes. Indeed, LANA1 alone, is thought to be sufficient for the maintenance, replication and segregation of terminal repeat (TR)-containing plasmids via binding of it's C-terminal domain to the TR sequence of the plasmids, which are then tethered to cellular chromosomes via it's N-terminal domain (4, 5, 15, 21, 24, 27, 51, 63). The relationship between chromosome tethering and other known LANA1 functions, such as regulation of gene expression, has not been clearly elaborated.
The pivotal role of LANA 1 in the life cycle of KSHV makes it a desirable target for antiviral therapy. The inability to generate large amounts of virions makes the development of vaccines for KSHV composed of killed viruses impractical. This failure underscores the importance of targeting cells latently infected with KSHV for use in antiviral therapies.
Intrabodies in a format of single-chain variable region antibody fragments can be expressed intracellularly and can bind to viral proteins, and other targets interfering with biological processes that take place inside the cell, representing a valuable tool in functional genomics at the protein level (ex. proteomics). Compared to other gene manipulation methods, such as gene knock-out or RNA-based technologies (ribozyme, antisense RNA or RNA interference), intrabodies have the unique advantage of targeting proteins in different cellular compartments as well as specific structural or functional motifs of a protein. Several LANA1 specific chimeric Fab antibodies from immunized rabbits were constructed by phage display technology, and converted to single chain antibodies which were linked through a peptidic bridge allowing the correct folding in sufficient amounts to be active as cytoplasmic intrabodies. The genetic approach described inhibited viral latency by interaction of the scFvs with LANA1, thereby inhibiting the segregation of KSHV DNA to daughter cells (14).
While progress has been made in specifically treating lytic herpesviral infections using viral DNA pol targeting, there is no effective therapy to target latent KSHV infection. RNAi approaches to inhibiting KSHV latency have been described which also show therapeutic promise (26). Surprisingly, RNAi against LANA1 is ineffective, although the authors demonstrated that the amount of LANA1 by western blot analysis was diminished. It is likely that this is due to the prolonged half-life of LANA1 protein (unpublished data, R. Sarid, Y. Chang) allowing even small amounts of LANA1 mRNA to code for sufficient protein to allow maintenance of the viral episome.
Upon interaction with the protein target, intrabodies are unique in their ability to modulate target protein function and/or achieve phenotypic/functional knock-out by several different mechanisms including accelerating target protein degradation and sequestering the target protein in a non-physiologic sub-cellular compartment. Innovative selection methods are now being used including array screening for high-avidity antibodies (17) and recovery of internalized phage from live cells to select against internalizing (human) receptors (53). Phage technology has been applied to complete proteome analysis using membrane-based screening (41). Other mechanisms of intrabody mediated gene inactivation are more dependent on the actual epitope to which the intrabody is directed, such as binding to the catalytic site on a target protein or to epitopes that are involved in critical protein-protein, protein-DNA or protein-RNA interactions. In this way, intrabodies have several advantages over gene inactivation strategies that globally disrupt target gene transcription (ex. antisense, ribozymes, siRNA) and generally do not dissect these type of molecular interactions (62, 70). In addition, by further exploiting their potential to be expressed in a spatially and temporally controlled manner through the use of inducible expression, for ex. of lentiviral or adenoviral (72) vectors, the use of these intrabodies as gene therapy in the future may be realized.
RNA interference is also being exploited to treat or prevent infection, and as a therapeutic against cancer. One use of RNAi is based on injecting large quantities of synthetic double stranded RNA (dsRNA) or DNA encoding short hairpin RNA (shRNA) intravenously or by transfection, which is not realistic for the treatment of human disease. The efficient delivery of therapies that knock-down specific RNA remains an obstacle to translate RNAi into a realistic treatment for human disease. Antisense RNA provides less inhibition of gene expression compared to RNAi, but the direct delivery of antisense RNA to treat CMV-retinitis has already been applied clinically with success. The incorporation of these shRNA or RNAi into lentiviral or adenoviral vectors offers the opportunity to use these vectors to target RNA efficiently in vivo.
However, the use of lentiviral delivered RNA interference to inhibit the multi-functional proteins like LANA1, opposed to other proteins maybe appropriate only in specific circumstances. RNAi is a phenomenon allowing sequence specific targeting and silencing of exogenous and endogenous gene expression. We have shown by mapping of LANA1 that several epitopes are recognized by the anti-LANA1 BM9 and BM10 that are effective in neutralizing LANA1's chromosome binding function. Others have shown that LANA1's turnover is very low (Patrick Moore, unpublished observation). Taking these two facts into consideration, we can conclude that RNAi inactivation of LANA1 would not be as effective as intrabody strategies, as LANA1 is expressed during a long time. This is confirmed by the results obtained by Godfrey et al. (26). The effectiveness of these relatively low binding affinity scFv in inhibiting latent KSHV infection provides a proof-of-principle for the feasibility of using engineered intracellular antibodies as a novel therapy against latent viral infections. Intrabody fragments could be delivered through gene therapy allowing direct treatment of latently-infected cells. In situ lysis of latently infected cells may provoke a protective immune response against viral antigens that could extend the efficacy of this form of treatment. The development of high-affinity anti-LANA1 scFv antibodies might result in even more efficacious anti-KSHV agents.