
Abstract
Almost 7 million children under the age 5 die each year, and most of these deaths are attributable to vaccine-preventable infections. Young infants respond poorly to infections and vaccines. In particular, dendritic cells secrete less IL-12 and IL-18, CD8pos T cells and NK cells have defective cytolysis and cytokine production, and CD4pos T cell responses tend to bias towards a Th2 phenotype and promotion of regulatory T cells (Tregs). The basis for these differences is not well understood and may be in part explained by epigenetic differences, as well as immaturity of the infant's immune system. Here we present a third possibility, which involves active suppression by immune regulatory cells and place in context the immune suppressive pathways of mesenchymal stromal cells (MSC), myeloid-derived suppressor cells (MDSC), CD5pos B cells, and Tregs. The immune pathways that these immune regulatory cells inhibit are similar to those that are defective in the infant. Therefore, the immune deficiencies seen in infants could be explained, in part, by active suppressive cells, indicating potential new avenues for intervention.
Introduction
Overview of Infant Mortality Rates Due to Infection
Infant mortality is a major global health problem, resulting in the commitment of 189 heads of state to achieve Millennium Development Goal (MDG) 4: to reduce the under-5 mortality rate by 2/3 between 1990 and 2015. Despite progress towards this goal, it is estimated that approximately 7 million children died before the age of 5 years in 2011; 1 almost 2/3 of these deaths were due to infection. The overwhelming majority of under-5 deaths are in Sub-Saharan Africa, which has an infant mortality rate (102 deaths per 1,000 live births) that is approximately 15 times higher than in developing countries. 2 The 3 infections that cause the most substantial morbidity and mortality are human immunodeficiency virus (HIV), tuberculosis (TB) and malaria, and all 3 of these infections disproportionately affect young children.3–5
Neonates and young children are known to respond poorly to infection. For example, neonatal and infant HSV- and CMV-specific CD4pos T cell responses are delayed and reduced as compared to adults with primary infection.6–10 Also, young infants are more likely to die or be hospitalized if infected with pertussis, respiratory syncytial virus (RSV), Enterovirus 71 or influenza than older children are.11–15 Furthermore, infants respond sub-optimally to most vaccines (reviewed in16–18). Sub-optimal responses to vaccinations and infection have traditionally been attributed to a combination of the immaturity of the neonatal/infant immune system and interference from passively transferred maternal antibodies. Here we will discuss data in support of a third potential mechanism: active suppression of early life responses.
Overview of Poor Response Rates to Vaccination
Infants are notorious for their reduced ability to respond to vaccination (reviewed in16–18), which is exacerbated by concomitant parasitic infection (reviewed in 19 ). Antibody responses can be classified as T-cell-independent (TI) or T-cell-dependent (TD). In humans, TI responses do not reach adult levels until around 4–5 years and are absent before 3 months of age.20,21 TD responses are present early in life and can be induced by infection in utero. However, multiple immunizations are required in young infants in order to obtain protective immunity and these responses often wane without subsequent booster immunizations (reviewed in 20 ). The reasons for this reduced ability to induce and maintain protective antibody titers are just beginning to be elucidated and limitations of T cell function early in life are likely contributors. Although infants are capable of mounting Th1 and CD8pos CTL responses to infection, the majority of vaccine-induced CD4pos T cell responses appear to be Th2-biased.22–25 Furthermore, upon stimulation through the TCR, CD4pos T cells will preferentially differentiate into Tregs rather than effector CD4pos T cells. 26 However, this may be related to the type of antigens (mainly protein antigens)/adjuvants used for vaccination as live attenuated vaccines, such as bacillus Calmette-Guerin (BCG), which induce robust Th1 responses and CD8pos CTL responses in infants.27–31 It should be noted, though, that recent data show that cytokine profiles elicited by BCG vaccine can differ, dependent on the geographical region in which the infants reside.32,33 In addition, the mode of vaccine delivery may be important in determining Th bias, as use of attenuated Listeria monocytogenes as a vehicle to introduce antigen into the cytoplasm of APC has been shown to prime robust Th1 and CD8pos CTL responses in neonatal mice. 34 Naïve T cells require dendritic cells (DC) for efficient priming. Of note, after stimulation, neonatal myeloid DC do not up-regulate as much CD80 or CD40 as adult DC,35,36 suggesting that they are inferior at providing sufficient co-stimulatory signals for both T cells and B cells. As a consequence of defective nucleosome remodeling, 37 neonatal DCs also secrete less IL-12,24,38–40 which is required for both Th1 development and adequate NK cell responses.
Infant Immune Cell Immaturity
The following section summarizes some key observations regarding immaturity of the cellular immune response in neonates and young children. For a more extensive discussion of these developmental differences the reader is referred to a recent comprehensive review.18,41 Our review will highlight only a few aspects that we believe are important in the context of how infants respond to vaccination.
Dendritic Cells
The neonatal immune system is Th2-biased due to an epigenetic predisposition for enhanced IL-4 and IL-13 production, as well as a delayed maturation of both IL-12- and type I IFN-producing dendritic cells (reviewed in24,42). It has been consistently shown that neonatal conventional DCs secrete less IL-12 and plasmacytoid DCs secrete less type I IFN in response to TLR stimulation. 39 There is also a reduced ability to secrete IL-18, which acts in concert with IL-12 and type I IFN to activate NK cells. 43 However, secretion of IL-1beta, IL-6, IL-23 and IL-10 is similar to or even higher than adult levels, 38 suggesting that neonatal DCs do have the capacity to secrete cytokines but that their responses to stimulation differ from those of adults. Of note, combined TLR receptor stimulation appears to overcome the inability of neonatal DC to secrete IL-12, 44 which has potential implications for enhancing infant vaccination efficacy.
Natural Killer Cells
Natural killer (NK) cells are lymphocytes that control initial infection through cytokine production and the killing of infected cells in an MHC-independent manner without prior sensitization. 45 , 46 NK cells from umbilical cord blood consistently demonstrate poor cytotoxic function and generate reduced quantities of IFNγ and other cytokines when compared with NK cells obtained from adults (reviewed in47,48). We have demonstrated that cord blood contains increased frequencies of CD56 negative (CD56neg) NK cells with reduced expression of granzyme B and reduced production of IFNγ and the CC-class chemokines RAN-TES, MIP1α and MIP1β upon stimulation. 49 Both CD56pos and CD56neg NK subpopulations showed impaired viral suppression in cord blood, with impairment most marked in the CD56neg subset. This NK cell subpopulation may reflect an immature NK cell subset, as has been suggested previously. 50 Indeed, Gaddy et al have shown that incubation of CD56neg NK subpopulations with cytokines such as IL-12 and IL-15 matures these NK into adult-like cells with enhanced lytic ability. 50 NK cell survival, proliferation and cytotoxicity are dependent on numerous cytokines including IL-12, IL-15 and IL-18. The reduced ability of neonatal DC to secrete IL-12 and IL-18 may account for the reduced maturation of NK cells and result in the accumulation of immature CD56neg NK subpopulations with impaired viral suppressive activity. It should be noted that there are conflicting data regarding the level of Granzyme B expression and the ability of neonatal NK cells to perform cytotoxicity, leading some groups to suggest that neonatal NK cells are not simply immature versions of adult NK cells. 41 However, many of these other studies assessed NK cell activity in bulk populations (rather than separating the NK subpopulations) and used non-physiologic target cells (e.g., MHC-deficient K562 cells instead of autologous infected cells) making it difficult to assess how each subset contributes to lytic activity.
T Cells
Neonatal CD4pos T cells secrete equivalent amounts of IL-2 and proliferate in vitro as well as in adult cells in response to strong stimuli, such as anti-CD3/anti-CD28 ( 51 and Gervassi et al., submitted). However, there is evidence that proliferative responses to physiologic stimuli may be more limited (reviewed in 41 ). Unlike their ability to secrete IL-2, neonatal CD4pos T cells have a markedly reduced ability to secrete other cytokines, including IL-4, IL-5, and IFN-γ, among others.52,53 It is likely that the reduced ability of T cells to secrete cytokines is due to sub-optimal signal transduction events54–56 and to the fact that IFN-γ and IL-4 are only expressed in effector/memory cells, which are in low abundance in cord blood. 57 Notably, immature DC, and in particular reduced IL-12 production, may be responsible for the limited ability of neonatal T cells to produce IFN-γ. This premise is supported by the fact that we have shown that short-term addition of IL-7 and IL-12 can enhance IFN-γ secretion in response to polyclonal stimulation (Gervassi et al., submitted). Additional evidence for a lack of adequate support from neonatal DC comes from a study that showed decreased measles-specific T cell proliferation in infants after vaccination when compared to older children or adults, but enhanced T cell proliferation and IFN-γ secretion when infant PBMC were stimulated in the presence of exogenous IL-12 and IL-15. 58 There are multiple studies showing that neonatal T cells appear prone to differentiating into Th2 cells, rather than Th1 cells (reviewed in 41 ). Indeed, higher antibody and Th2 CD4pos T cell memory responses are induced in neonates compared to adults. 25 However, most studies used non-physiologic stimulation to activate neonatal T cells and such a skewing has not been observed when neonatal T cells are stimulated with allogeneic DC. 59
Neonatal T cells also have reduced cytolytic ability.60,61 There are conflicting reports as to the level of perforin expressed in neonatal CD8pos T cells;62,63 however, there are data suggesting that perforin expression is tightly correlated with proliferative ability 64 and thus it is plausible that neonatal T cells would express less perforin due to their limited proliferative ability.
B Cells
The timeline of B cell response maturation differs depending on the requirement for cognate T cell help. Humoral responses to polysaccharide antigens (such as capsular polysaccharides from Haemophilus influenzae type b, Neisseria meningitis, or Streptococcus pneumonia) do not require T cell help (TI) and are mediated by IgMpos IgDpos CD27pos splenic marginal zone B cells. These responses are triggered by crosslinking of the Ig molecules on the B cell surface along with cytokines produced by marginal zone antigen presenting cell (APC) such as B cell-activating factor belonging to the TNF family (BAFF) and A proliferation-inducing ligand (APRIL). 65 Newborns are exquisitely susceptible to infections and sepsis caused by these encapsulated bacteria and are non-responsive to polysaccharide vaccine antigens, due to a profound deficiency of TI B cell responses. These responses are undetectable before 3 months of age and do not fully develop until 4–5 years of age, partly due to the lack of maturity of the splenic marginal zone architecture66,67 and low expression levels of CD21. 68 These deficiencies can be partially overcome by signaling through TLR9, leading to emigrant B cells acquiring the CD27pos IgMpos phenotype and differentiation into IgM producing plasma cells. 69 Unlike TI B cell responses, TD B cell responses are present at birth, though of a lower magnitude when compared to older children or adults. These B cell responses are directed to protein antigens and are primed in the germinal centers with the help of follicular dendritic cells and follicular T helper cells. Lower TD responses could be attributable to the lower expression of CD40 ligand on CD4pos T cells,36,70 the delay in maturation of the follicular dendritic cell network, 71 decreased somatic hypermutation limiting affinity maturation of antibodies,72,73 an inability for long-lived plasma cells to establish themselves in the bone marrow due to a lack of stromal cell support,74,75 and an overall inability of neonatal dendritic cells to prime effective T helper responses (see above). Vaccination to protein or conjugate antigens does prime protective B cell responses; however, the magnitude and duration of these responses is significantly lower compared to older children or adults, necessitating several boosters (reviewed in 21 ). Nevertheless, some of the neonatal deficiencies that limit effective T-dependent B cell responses may also be circumvented by the use of specific adjuvants with the acquisition of adult-like B cell responses. 76
Infant Immune Cell Suppression
Successful pregnancy relies on the establishment of a variety of immune-regulatory mechanisms to prevent HLA-mismatched inflammatory responses between fetus and mother. Bilateral transfer of nucleated cells occurs between the fetus and the mother during pregnancy,77,78 with the establishment of fetal-specific polyfunctional T cell responses. 79 The establishment of several tolerance mechanisms to protect the fetus have been identified in the pregnant mother, and failure to establish tolerance has been associated with miscarriage and fetal resorption.80–84 Some of these maternal mechanisms include regulatory T cells,85–87 regulatory NK cells,88,89 regulatory dendritic cells, 90 epigenetic silencing of T cell-attracting chemokines, 91 indoleamine 2,3-dioxygenase (IDO),84,92 immunoregulatory hormones93–96 and regulatory molecule expression such as Human leukocyte antigen G (HLAG), 90 galectin-1,97,98 CD137 (4-1BB), 99 ICOS-L (B7h), 100 PD1/PDL1,101–103 and Tim3. 104 Less is known of the establishment of such mechanisms in the fetus, how these extend into neonatal/infancy and how they may modulate neonatal immune responses. Neonates can develop adult-like T cell responses under appropriate conditions (reviewed in 105 ), and the inability to mount effective responses in infants may be due to the presence of specific suppressive mechanisms. Here we provide a summary of the fetal immune-suppressive mechanisms described thus far, their persistence into infancy and their potential to modulate vaccine-specific immune responses.
Regulatory T Cells (Tregs)
Tregs have been shown to change in utero in the developing fetus in response to maternal alloantigens.78,106,107 Treg levels decrease with gestational age and attain adult-like levels by birth ( 107 and our unpublished data). Notably, these studies looked at bulk frequencies of Tregs and not whether antigen-specific subsets persist at elevated levels. Neonatal Tregs are potent suppressors of neonatal allogeneic T cell proliferative responses and depletion of these cells restores strong allo-proliferation.78,106,107 Neonatal Tregs also exert strong suppression of P. falciparum-specific T cell proliferation and IFN-γ production in infants exposed to malaria in utero. 108 This suggests that neonatal T cells are functionally capable of proliferation and production of IFN-γ in response to specific antigenic stimulation, and that these responses are actively suppressed.
CD5pos B Cells (B-1 Cells)
CD5pos B cells comprise about 40% of B cells at birth, increasing during the first 4 months of life, after which they gradually decrease.109–111 They are an important source of low affinity IgM and have been suggested to be a first line of defense against infection.111,112 CD5pos B cells also have an immune-regulatory function and have been shown to skew the T cell response towards a Th2 phenotype in neonatal murine models. This effect is mediated by secretion of IL-10 by CD5pos B cells upon TLR stimulation and suppression of IL-12 production by dendritic cells. In these studies, purified neonatal DCs were shown to have the capacity to produce IL-12 and type-1 IFN in response to TLR engagement, 113 but these responses were inhibited by CD5pos B cells. CD5pos T cells are also increased in neonates exposed to HIV with a concomitant increase in IL-10 production by mononuclear cells. 114
Myeloid-Derived Suppressor Cells (MDSC)
MDSC are a heterogeneous population of immature myeloid cells with suppressive function (reviewed in115,116). They can be characterized into monocytic-like (M-MDSC) or granulocytic-like (G-MDSC), dependent on specific surface phenotypes. In humans, M-MDSC are most often classified as CD33pos, CD11bpos, HLA-DRneg, and CD14lo/pos, whereas G-MDSC are CD33pos, CD11bpos, HLA-DRneg, CD14neg, and CD15pos, although many different marker combinations have been used (reviewed in 117 ). MDSC are normally present at low frequency in healthy individuals but their frequencies increase in situations of persistent inflammation, such as chronic infection,118,119 autoimmunity 120 and malignancy (reviewed in 121 ). MDSC suppress T cell activation using a variety of mechanisms (reviewed in117,1 2 2 ) and have been shown to decrease efficacy of dendritic cell vaccines. 123 MDSC have also been shown (in cancer models) to skew immunity to a Th2 response, 124 which is a well-known characteristic of the infant immune response. Recently, our group and others 125 have demonstrated that healthy neonates possess increased frequencies of G-MDSC. These neonatal MDSC potently suppress both T cell proliferation and IL-5, IL-17 and IFN-γ cytokine secretion ( 125 and Gervassi et al; submitted). Of note, we have observed elevated levels of G-MDSC up to 6-weeks of age and the increase in the frequency of these cells cannot be attributable to the transient increase in granulocytes that occurs at birth.
MDSC also inhibit NK cell cytotoxicity, IFN-γ secretion and NKG2D expression in mouse cancer models, 126 raising the intriguing possibility that they may also suppress NK cell function in human neonates.
Mesenchymal Stromal Cells (MSC)
MSC are multi-potent cells that can be isolated from almost every postnatal organ and tissue 127 and are capable of differentiating into several mesodermal, endodermal and ectodermal lineages. 128 They have gained much attention as tissue regenerative therapy for their self-renewal and differentiation capabilities and because they do not express MHC class II or CD40, CD80 or CD86 costimulatory molecules. These cells also inhibit a variety of innate and adaptive immune responses and are currently under clinical investigation/use to prevent graft failure, to promote hematopoietic stem cell transplantation and as treatment for certain inflammatory and auto-immune conditions.
MSC have not been investigated as potential immunemodulatory cells during gestation or infancy and to our knowledge there are no studies looking at the temporal frequency or specific location of these cells in infants. MSC are present at a high frequency in cord blood from gestational weeks 24–28, greatly decrease by gestational weeks 29–32 and are rarely identified in gestational weeks 37–40. 129 They are found in full-term placental tissue, fetal membrane, and neonatal and infant thymus.130–132 MSC have multi-faceted immunomodulatory effects. They inhibit differentiation of monocytes into immature DC and differentiate DC into regulatory DC and M-MDSC.133–135 Regulatory DC, along with MSC, induce functional Foxp3pos Tregs from committed Th1 and Th17 cells.133,136–138 Furthermore, MSC skew the T helper response towards a Th2 phenotype, 139 suppress the lytic activity of CD8pos T cells, 140 suppress proliferation and induce anergy of both naïve and memory T cells.141,14 2 They suppress proliferation, cytotoxicity and cytokine production by NK cells.139,143,144 These immune regulatory effects are mediated by the production of prostaglandin E2 (PGE2), HLA-G, TGF-beta, indoleamine 2,3-dioxygenase (IDO), IL-10 and IL-6.134,136,139,144–147
Conclusions and Potential for Intervention
It will be critical to determine if Tregs, MDSC, MSC, CD5 B cells, or other regulatory cells are responsible for the reduced ability of infants to respond to vaccination/infection early in life. Observational studies aimed at addressing this issue are already underway. There is a possibility that regulatory cells could have beneficial effects, such as limiting inflammation induced during colonization of the human gut with commensal microbiota. However, the extreme heterogeneity in frequencies of MDSC that we have observed in healthy neonates argues that such a role may be limited.
Given the overwhelming data showing that the neonatal/young infant cellular immune system is immature and potentially actively suppressed, the use of novel adjuvants seems worth exploring. Many adjuvants are being specifically developed to stimulate through specific TLRs. However, the response of neonatal DC to TLR stimulation is very different than the responses seen in adults, as described above. 38 A recent review of the data on use of adjuvants in infants suggests that many adjuvants may be safe and well tolerated 148 but some, such as TLR2 agonists, may induce expression of IL-10 and lead to unresponsiveness to the vaccine. The use of TLR agonists as adjuvants may be attractive in infants because certain TLR agonists are known to inhibit the immunosuppressive function of MDSC (TLR 9 agonist CpG)131,149,150 and of MSC (TLR2 agonist Pam2Cys). 151 However, caution should be employed as other TLR agonists (such as the TLR 7 agonist, imiquimod) have been shown to induce MDSC accumulation. 152 There have been no studies to date determining if administration of specific adjuvants can overcome the Th2-bias of most protein-based vaccines antigens. Thus, more studies are required to assess the potential adjuvant effects of different TLR agonists. However, obviously, extensive safety monitoring will be required to assess potentially damaging inflammatory sequelae.
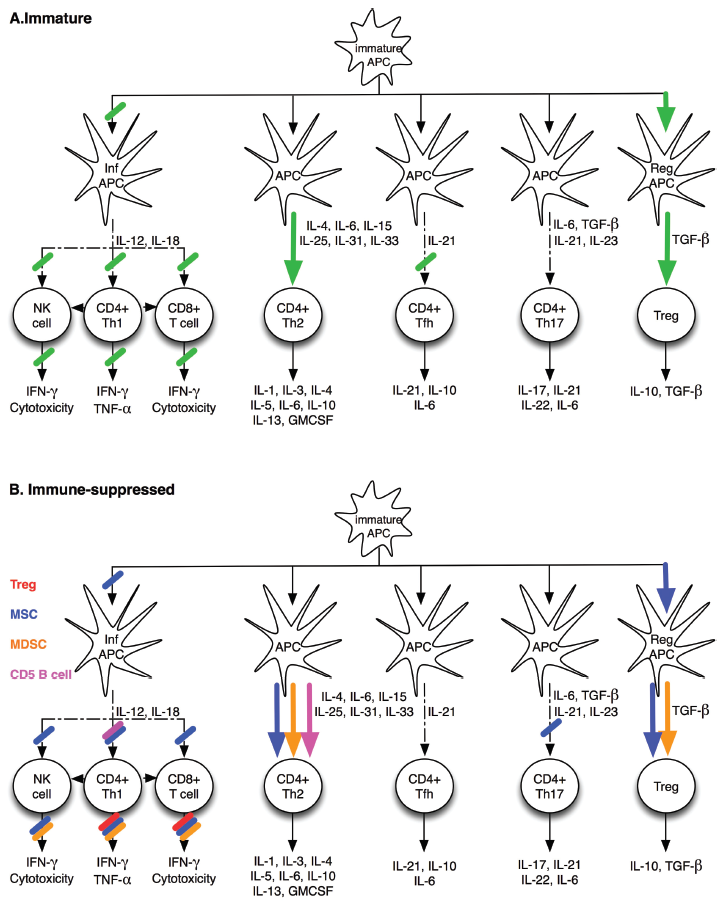
Known characteristics of the infant's immune responses.
An alternative strategy is to use vitamin A supplementation at an earlier age than is currently recommended (6 months 153 ). Vitamin A supplementation has been shown to reduce mortality and morbidity from some forms of diarrhea, measles, human immunodeficiency virus (HIV) infection, and malaria. 154 The mechanism of its efficacy is not known but it is interesting that all-transretinoicacid (ATRA), the active metabolite of vitamin A, causes maturation of MDSC and improves response to vaccination and subsequent tumor regression in cancer patients,121,155–157 Thus, elimination of MDSC using vitamin A or ATRA may be a safe and effective means of enhancing infant immune responses.
A final approach is BCG vaccination. BCG has been shown to increase Th1- and Th2-responses to other vaccines such as HBV and oral polio vaccine. 148 While this has been attributed to the potential of BCG to cause maturation of DC, 158 it is possible that the numerous TLR agonists in BCG cause maturation/elimination of MDSC or other regulatory cells, thereby relieving immune suppression.
In summary, there are extensive data supporting the role of active immune suppression during early infancy. This active suppression may be responsible for the reduced ability of infants to respond to infection/vaccination. New approaches aimed at alleviating such immune suppression may enable infants to respond more appropriately to infection/vaccination and speed progress towards MDG4.
Author Contributions
ALG and HH wrote the first draft of the manuscript. ALG and HH contributed to the writing of the manuscript. ALG and HH agree with manuscript conclusions. ALG and HH jointly developed the structure and arguments for the paper. ALG and HH made critical revisions and approved final version. ALG and HH reviewed and approved of the final manuscript.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests. Provenance: the authors were invited to submit this paper.