
Abstract
Persistent infections with human immunodeficiency virus type 1 (HIV-1) and hepatitis C virus (HCV) are a major cause of morbidity and mortality worldwide. As sentinels of our immune system, dendritic cells (DCs) play a central role in initiating and regulating a potent antiviral immune response. Recent advances in our understanding of the role of DCs during HIV-1 and HCV infection have provided crucial insights into the mechanisms employed by these viruses to impair DC functions in order to evade an effective immune response against them. Modulation of the immunological synapse between DC and T-cell, as well as dysregulation of the crosstalk between DCs and natural killer (NK) cells, are emerging as two crucial mechanisms. This review focuses on understanding the interaction of HIV-1 and HCV with DCs not only to understand the immunopathogenesis of chronic HIV-1 and HCV infection, but also to explore the possibilities of DC-based immunotherapeutic approaches against them. Host genetic makeup is known to play major roles in infection outcome and rate of disease progression, as well as response to anti-viral therapy in both HIV-1 and HCV-infected individuals. Therefore, we highlight the genetic variations that can potentially affect DC functions, especially in the setting of chronic viral infection. Altogether, we address if DCs’ potential as critical effectors of antiviral immune response could indeed be utilized to combat chronic infection with HIV-1 and HCV.
Keywords
Introduction
The immune response generated during a viral infection involves a complex interplay between the virus and the two arms of the immune system, innate and adaptive. Dendritic cells (DCs) are a specialized category of professional antigen-presenting cells (APCs) that act as messengers between the innate and the adaptive immune system. 1 Immature DCs are derived from hematopoietic bone marrow progenitor cells and are widely distributed within tissues such as the skin, mucosal surfaces, and blood that come in direct contact with the external environment. DCs are equipped with pattern recognition receptors (PRRs) such as Toll-like receptors (TLRs), whose role is to sense a wide array of pathogen-associated molecular patterns (PAMPs). In humans, the TLR family consists of 10 members, named TLR1-10, with each member being specific for the PAMP it recognizes; TLR7, for example, recognizes single-stranded RNA and TLR3 recognizes double-stranded RNA. 1 Plasmacytoid DCs (pDCs) express TLR7 and TLR9, whereas myeloid DCs (mDCs) express TLR1-3 and TLR8. 2 Upon TLR-mediated viral sensing, DCs get activated and migrate to lymph nodes where they prime a naive T cell against the viral peptide that is presented on their surface by MHC molecules. DCs can process both extracellular antigens via the lysosomal pathway and intracellular proteins via the proteasomal pathway. 3 After viral processing, DCs become activated and migrate to the draining lymph nodes, where they transform into mature DCs in the T-cell-rich areas. Maturation of DCs involves several changes including cytoskeleton reorganization, redistribution of MHC molecules from endocytic compartments to the surface, inhibition of antigen uptake, and an increase in the expression of co-stimulatory and adhesion molecules as well as chemokine receptors. 4
DCs exhibit heterogeneity at several levels including phenotype, function, and anatomical location. 5 DCs in the epidermis are referred to as Langerhans cells (LCs), dermal DCs are found in dermis, and interstitial DCs are found in all peripheral tissues except skin. Blood DCs in turn are broadly classified into two major groups, mDCs and pDCs, with mDCs being further comprised of different subsets. Table 1 summarizes the phenotype and functional characteristics of various DC subsets, clearly indicating a low frequency of DCs in blood. To facilitate ex vivo analysis of blood DCs, we have recently developed an antibody cocktail for polychromatic flow cytometry and evaluated its applicability for immune profiling of human T-cell leukemia virus type 1 (HTLV-1), as well as HIV-1/ HCV co-infected patient cohorts. These observations remain unpublished. We have also demonstrated the suitability of using this newly developed cocktail in immunological investigations of frozen peripheral blood mononuclear cells (PBMCs) from infected patients. The use of multi-parametric antibody cocktails has been proven to be very useful in assessing the frequency as well as phenotypic and functional changes on rare DC subsets during viral infections.
Frequency and phenotype of blood DC subsets.
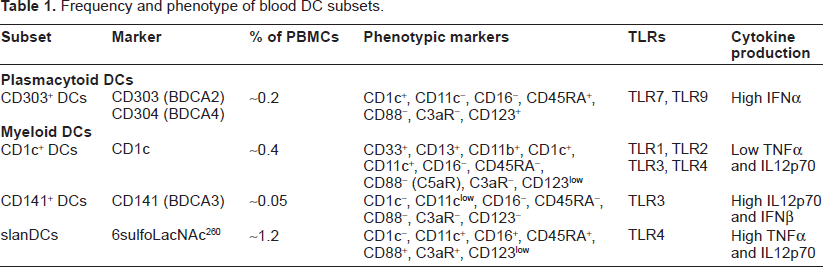
Different DC subsets have acquired both distinct and overlapping roles in the immune system. 6 Figure 1 illustrates the role of DCs in initiating and regulating the adaptive immune response. Activated mDCs are responsible for priming CD4+ and CD8+ T cells against a specific pathogenic epitope. In addition, they produce interleukin (IL)-12 and IL-15, both of which activate natural killer (NK) cells and promote the differentiation of epitope-specific CD4+ T-helper (TH) cells and CD8+ T cells into TH1 cells and cytotoxic T lymphocytes (CTLs) respectively. Activated NK cells have two major roles. The first role is to secrete interferon-γ (IFNγ), which inhibits viral replication in infected cells. Secondly, NK cells kill virus-infected cells directly through the release of cytolytic mediators such as granzyme. TH1 cells also secrete IFNγ, which stimulates the activation of CTLs and the production of neutralizing immunoglobulin G2a antibodies by plasma cells. In contrast to mDCs, whose main role is to prime T cells against the virus, pDCs function as one of the earliest mediators of antiviral immune response by producing large amounts of type I IFNs (IFNα and IFNβ) upon viral sensing. Besides inducing an antiviral state in virus-infected cells, type I IFNs take part in activation of NK cells 7 and promotion of CD4+ and CD8+ T-cell differentiation and survival. 8 In addition, pDCs have also been shown to act as APCs.9–11 Interestingly, recent data suggests that pDCs can facilitate CD4+ TH cell responses to persistent viruses independently of direct antigen presentation, 12 13 necessitating the need to assimilate enough knowledge about the role of pDCs in antiviral immune response.
Role of DCs in the antiviral immune response.
The importance of DCs in resolving viral infection has been shown for several viruses including human respiratory syncytial virus and influenza virus.14, 15 Our group is focused on gaining complete understanding of the role of DCs in chronic viral infections caused by HTLV-1, HIV-1, and HCV.16–28 Many studies including ours strongly suggest that DCs play an important role in the control of HCV and HIV infection, two of the most common and fatal blood-borne viruses. Patients in whom HCV infection clears spontaneously or in whom HIV-1 infection is controlled have strong multi epitope-specific CD4+ and CD8+ T-cell responses; these responses probably reflect the efficient antigen presentation and T-cell activation potential of DCs.29, 32 However, the chances of viral clearance after HCV and HIV-1 infection are low, owing to the remarkable ability of these viruses to evade the host immune response and establish chronic infection.
Chronic HIV-1 infection causes acquired immunodeficiency syndrome, 33 which is characterized by severe immunosuppression and associated opportunistic infections. 34 According to a recent report by the Joint United Nations Programme on HIV/AIDS, an estimated 33.4 million people around the world are living with HIV infection. 35 The Centers for Disease Control and Prevention (CDC) estimated that 1,178,350 persons aged 13 and older were living with HIV infection in the United States as of 2008. 36 In 2009, there were an estimated 48,100 new HIV infections. 37 Chronic HCV infection causes chronic liver disease and its associated complications such as liver cirrhosis, liver failure, and hepatocellular carcinoma. 38 According to the latest update by the World Health Organization (WHO), about 170 million people are chronically infected with HCV and more than 350,000 people worldwide die from HCV-related liver disease each year. 39 Figure 2 outlines the outcome and the course of disease progression during HCV infection. It also outlines important factors such as gender, age, ethnicity, and host genetic makeup that favor spontaneous clearance of HCV infection. Apart from HIV-1 and HCV mono-infected patients, a significant population of co-infected patients exists. Overall, co-infection with HCV is found in 30% of HIV-infected patients and in 60% to 90% of HIV-infected injection drug users.40–42
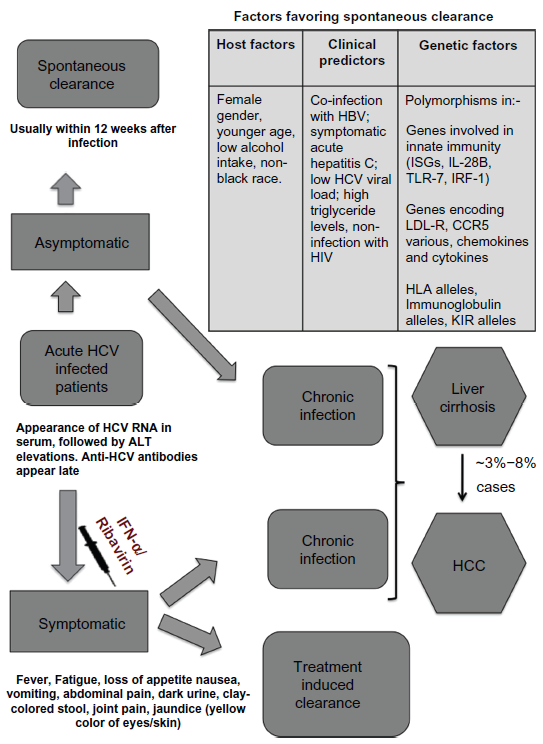
Outcome of acute HCV infection.
A weak and narrowly focused antiviral T-cell response is a common denominator in both HIV-1 and HCV infections. Being one of the key players involved in initiation and regulation of the antiviral immune response, DCs are the ideal targets for HIV-1 and HCV to exercise their immune evasion strategies. Therefore, in order to develop novel therapies against chronic HIV-1 and HCV infection, there is a need to understand the interaction between DCs and both these viruses. This review discusses the mechanisms by which HIV-1 and HCV are able to interfere with the normal immune response of the host. We also highlight the role of host genetic variations in modulating various DC functions associated with antiviral immunity. Finally, we discuss the recent advances in the field of anti-HCV drugs.
Do HIV-1 and HCV Infect DCs?
The presence of DCs in the skin and blood as well as at the mucosal surfaces, and their proficiency in antigen uptake, predisposes them to be the primary targets for viruses. Therefore, it is possible that HIV-1 and HCV infect DCs. Replication of the viral genome along with the expression of viral proteins in DCs might interfere with many signaling pathways in DCs, leading to impairment of DC functions and thus rendering them incapable of stimulating a proper T-cell response against the virus. For example, ICP47 of herpes simplex virus 1 and US6 of human herpes virus 5 are known to inhibit the loading of antigenic peptides onto MHC class I molecules, leading to a reduced ability of infected DCs to prime naïve T cells efficiently. 43
HIV-1
The main target cells for HIV-1 are mononuclear leukocytes bearing CD4 and the chemokine receptors CC-chemokine receptor 5 (CCR5) and CXC-chemokine receptor 4 (CXCR4);44–48 however, LCs, pDCs, and mDCs have also been reported to be susceptible to HIV-1 infection.49–61 HIV-1 replication is generally less productive in DCs than in CD4+ T cells,49,57,59,62–64 and the frequency of HIV-1-infected DCs in the blood has been reported to be 10 to 100 times lower than that of CD4+ T cells. 65 Immature DCs are more susceptible to HIV-1 infection than mature DCs, 66 perhaps partly as a result of their higher antigen uptake capacity. Interestingly, replication of HIV-1 in pDCs was found to increase substantially following CD40 ligation. 67 CD40 is a co-stimulatory molecule found on APCs and its binding to CD40 ligand (CD40L) on TH cells activates APCs to perform a variety of functions. Thus, HIV-1 replication in pDCs may be upregulated through its interaction with activated CD4+ T cells in the extrafollicular zones of the lymphoid tissue.
HCV
Hepatitis C virus primarily infects and replicates in hepatocytes; however, evidence is accumulating about extrahepatic replication of HCV, especially in PBMCs. The highly conserved 5’ untranslated region of HCV RNA has been shown to vary between PBMCs and plasma in the same individual,68,69 suggesting that HCV alters its replication inside non-hepatic cells. Lymphoid cells may act as HCV reservoirs, thus contributing to the establishment of chronic infection. Different studies have shown low levels of HCV RNA in serum and lymphoid cells after spontaneous or IFN/RBV-induced resolution of chronic hepatitis C.70,71 In patients with a sustained virological response, small quantities of HCV RNA have been shown to persist in the liver or in PBMCs for up to 9 years. 71 HCV may persist and replicate in the liver and PBMCs of healthy, anti-HCV antibody positive, serum HCV RNA-negative patients who have persistently normal alanine amino transferase levels. 72 This could present a potential risk for reactivation, especially in the case of immunosuppression due to the presence of HIV-1 infection in the same individual. In fact, HCV RNA in peripheral blood cell subsets in HIV/HCV co-infected patients at the end of IFN/RBV treatment is associated with virological relapse. 73 Interestingly, HCV RNA has been shown to reemerge in apparent sustained virological responders receiving immune suppressive therapy.74,75 This suggests that HCV persists years after IFN/RBV- induced resolution from plasma and suggests that continued immune surveillance is required to prevent its recurrence, even in sustained virological responders. 74 As far as DCs are concerned, HCV genomic RNA has been detected in mDCs and pDCs isolated from the blood of HCV-infected patients.76,77 One study, which used strand-specific semi quantitative reverse transcription polymerase chain reaction, has shown that 3 out of 24 HCV-infected patients had DCs carrying the replicative intermediate of HCV. 76 More such studies are needed to clearly establish whether HCV replicates in blood DCs of infected patients. In one study, monocyte-derived DCs (MDDCs) from healthy donors were found to contain HCV replicative intermediate when incubated with the serum of HCV-infected patients. 78 This study provides evidence that DCs can at least support the first steps of the viral life cycle. However, other studies wherein MDDCs and various blood DC subsets were incubated with infectious recombinant HCV were not successful in detecting viral replication or viral protein synthesis.79, 81
In summary, DCs can support HIV-1 replication, although at a level much lower than CD4+ cells (primary targets for HIV-1); additionally, they cannot support the production of HCV virions, even though HCV may be able to infect DCs and initiate its replication. Many explanations are possible. Firstly, as professional APCs, DCs might degrade the virus in intracellular compartments before it completes its replicative cycle. DC-specific ICAM-3 (intercellular adhesion molecule-3) grabbing non integrin (DC-SIGN or CD209) is a C-type lectin receptor (CLR)82,83 that has been shown to promote uptake and presentation of HIV-1 by MHC class I and class II molecules. 84 Scavenger receptor class B member 1 (SRB1) has been shown to mediate in HCV uptake and presentation. 85 The second possibly explanation involves the fact that host factors required for HIV-1 and HCV replication might be missing in DCs, or DC-specific host factors might restrict HIV-1 and HCV replication. For example, cellular factors such as cytidine deaminase APOBEC3G (apolipoprotein-B mRNA-editing enzyme, catalytic polypeptide-like 3G),86,87 the transcription factor NFAT (nuclear factor of activated T cells), 88 the nuclear factor-κB (NF-κB) regulating protein (MURR1), 89 and polypyrimidinetract-binding protein (PTB) 90 have been shown to block HIV-1 replication after its entry into resting CD4+ T cells, although the relevance of these cellular restriction factors in DCs is not fully studied. Expression levels of APOBEC3G have been shown to correlate with resistance of mDCs to HIV-1. 91 APOBEC3G upregulation by IFNα has been shown to restrict HIV-1 infection in pDCs. 92 Additionally, engagement of the receptors CCR5 and CD40 with CCL3 and CD40L respectively enhances the expression of APOBEC3G in DCs through activation of the ERK 1/2 and p38 MAP kinase signaling pathways. The third possible explanation is that DCs express low levels of HIV-1 receptor CD4, as well as co-receptors CCR5 and CXCR4. 93 HCV co-receptor claudin1 is also present at low levels in DCs. 81 HIV-1 and HCV entry receptors are reviewed in the next section.
HIV-1 uptake by DCs has been shown to trigger TLR8 (via ssRNA), which results in phosphorylation of RNA Polymerase II (RNAPII) at serine 2 of the C-terminal domain (CTD). 92 On the other hand, binding of HIV-1 envelope protein gp120 with DC-SIGN results in the activation of Raf-1 and ultimately leads to phosphorylation of p65 at serine 276. This modification on p65 recruits positive transcription elongation factor b (p-TEFb) to the long terminal repeat (LTR), which phosphorylates RNAPII at serine 5 of CTD. 92 It has been shown that phosphorylation of RNAPII at both serine 2 and serine 5 of its CTD promotes full-length transcription of the integrated HIV-1 genome and production of HIV-1 proteins. Thus, HIV-1 induces innate signaling by TLR8, as well as DC-SIGN, and that allows it to cause productive infection of DCs. Despite this, the frequency of blood DCs productively infected with HIV-1 is low. This could be due to increased migration of these DCs from blood to lymph nodes or due to increased rate of apoptosis. Interestingly, HCV glycoprotein E2 is known to interact with DC-SIGN 94 and this interaction has been shown to activate p38 MAPK pathway, possibly leading to activation of Raf-1. Therefore, it would be interesting to study whether HCV can augment HIV-1 replication in DCs during HIV-1/HCV co-infection.
Role of DCs in Viral Dissemination
Following antigen uptake and activation, DCs present in peripheral tissues migrate to secondary lymphoid organs and present the antigen to the naive T cells. It is therefore not unreasonable to propose that various viruses would exploit this migratory property of DCs in order to disseminate to sites that are favorable for their replication. Herein, we review the entry and recognition of HIV-1 and HCV into DCs.
HIV-1
As some of the first target cells that encounter HIV-1 at the mucosa, DCs contribute to the initial stages of HIV-1 infection and dissemination.95,96 HIV-1 has been shown to inhibit the fusion between endosomes and autophagosomes, which not only leads to prevention of autophagy-mediated viral degradation and subsequent presentation of viral antigens, but also enhances the survival of HIV-1 inside DCs.97–100 Enhanced survival of HIV-1 is likely to promote its dissemination to T cells by various mechanisms. Binding and internalization of HIV-1 by DCs is mediated by HIV-1 entry receptor CD4 and co-receptors CCR5 and CXCR4, which are known to be expressed on the surface of DCs.101,102 Other chemokine receptors that are also suggested to participate in HIV-1 entry are CCR3, CCR8, CCR9, and CXCR6.96,103 In addition, DCs express C-type lectins such as DC-SIGN, langerin (also known as CLEC4K or CD207), and DC immunoreceptor (DCIR; also known as CLEC4A), which bind to HIV-1.96,104 Expression of these receptors depends on the DC subtype, activation state, and localization. Even though pDCs express C-type lectins such as BDCA2, binding of HIV-1 to pDCs is generally mediated by interaction of HIV-1 envelope protein gp120, with CD4 expressed on pDCs. Interestingly, the identity of receptors involved in virus-DC interaction seems to affect the fate of virus after its binding to the cell surface. The interaction between the carbohydrate recognition domain of DC-SIGN and gp120 results in internalization of HIV-1 into non-lysosomal compartments in DCs, where it does not undergo degradation. The internalized virion can then be transported within DCs before finally getting transferred to T cells in a process known as transinfection. This model of DC-dependent infection of CD4+ T cells, commonly referred to as the Trojan horse model, involves endocytosis of HIV-1 virion by DCs followed by its transmission to the T cells via vesicular exocytosis across the infectious synapse. This structure is analogous to the immunological synapse formed between DCs and T cells in the T-cell areas of lymphoid tissue. 105 A conflicting study, 106 however, demonstrated that transinfected HIV-1 was fully sensitive to surface-applied soluble CD4, which has been shown to inactivate HIV-1 and inhibit its entry even after the virus was sequestered within the DCs. This phenomenon suggests that the sequestered, internalized HIV-1 virions are targeted for degradation and only the surface-bound virions participate in transinfection. A careful reexamination of these observations in a different study 107 showed that mature DCs concentrate viral particles within membrane folds, making them accessible for surface-applied soluble CD4. This suggests that soluble surface-applied CD4-induced reduction in transinfection could be due to inactivation of sequestered HIV-1 virions and that sequestered, internalized HIV-1 could participate in transinfection.
The binding of HIV-1 to DC-SIGN enhances the formation of infectious synapse; the engagement of DC-SIGN by HIV-1 activates Cdc42 and induces membrane extensions in immature DCs, thus facilitating viral propagation across the infectious synapse. It has also been observed that CD4+ T cells participate in transinfection by extending membrane protrusions into the virus-containing pockets on DCs. This helps the virus to emerge from the pocket by engaging CD4 on the membrane protrusions. DC-SIGN-independent mechanisms are also involved in DC-mediated HIV-1 transinfection of CD4+ T cells.108–113 Recent data indicates that immature DC-mediated HIV-1 transinfection is partially dependent on DC-SIGN, whereas mature DC-mediated HIV-1 transmission to different target cells is independent of DC-SIGN and C-type lectins. 114 However, another study has shown why interaction of HIV-1 with DC-SIGN may be important for efficient HIV-1 transmission. Clusterin (an apolipoprotein) in semen is differentially glycosylated compared to clusterin in blood. Clusterin in semen is a DC-SIGN ligand and can therefore block the interaction between HIV-1 and DC-SIGN, thereby preventing HIV-1 transmission to CD4+ T cells. This may also explain why exposure to HIV-1 through sexual contact is less likely to result in infection compared to blood-borne exposure. 115
It is known that maturation of DCs increases their efficiency in transmitting HIV.56,106,107,114,116–121 DC maturation leads to an upregulation in ICAM-1 (CD54) gene expression, which correlates with mature DC-enhanced transmission of HIV-1 to CD4+ T cells. 120 Interactions between ICAMs and their ligands on T cells can thus be proposed to facilitate DC-T-cell contact and HIV-1 transmission. In fact, it has been shown that ICAM-1 (but not ICAM-2 or −3) is important for DC-mediated HIV-1 transmission 122 and that blocking ICAM-1 on DCs impairs HIV-1 transmission. 120 Because the interaction between ICAM-1 and leukocyte function-associated molecule 1 (LFA-1) is important for DC-T-cell adhesion, 123 it has been proposed that the interaction between ICAM-1 on DCs and LFA-1 on CD4+ T cells is important for HIV-1 transinfection. 120 As mentioned previously, HIV-1 can also infect DCs, although less efficiently than CD4+ T cells. Many reports have indicated that infected DCs can aid in HIV-1 dissemination by transmitting the viral progeny to T cells; this process is known as cisinfection.121,124 Interestingly, HIV-1-infected MDDCs show Nef-induced downregulation of CD4 that correlates with enhanced viral transmission. 125 These results suggest that CD4, which is present at various levels in DC-SIGN-positive primary cells, could be one of the key regulators of HIV-1 transmission.
It is also important to understand the role of different DC subsets in HIV-1 transmission. pDCs are less efficient than mDCs in viral transmission. 126 LCs also appear to transinfect HIV-1 through surface exposed structures similar to those in mature DCs.106,116 However, one study suggested that expression of CLR langerin on LCs acts as a barrier to HIV-1 transmission. 127 Because different mDC subsets have been shown to transfer HIV-1 to activated CD4+ T cells, 96 the question of whether LCs can do so needs to be resolved.
HCV
The idea that DCs might play an important role in dissemination of HCV, a hepatotropic virus, is still in its infancy. HCV envelope protein E2, serum of HCV-infected patients, and retroviruses that are pseudotyped with HCV envelope proteins have all been shown to bind specifically to DC-SIGN.94,128,129 Therefore, it is possible that DC-SIGN mediates the binding of blood DCs or hepatic DCs in liver sinusoids to the circulating HCV particles. MDDCs expressing DC-SIGN were shown to bind HCV pseudovirus and transmit it efficiently when co-cultured with the human hepatocellular carcinoma cell line Huh7; Huh71 is a cell line that is widely used to study HCV pseudovirus entry as well as productive replication of recombinant infectious HCV.129,130 Importantly, HCV virus-like particles were shown to bind DC-SIGN and get targeted to nonlysosomal compartments within MDDCs. These particles were thus protected from lysosomal degradation, suggesting that, similar to HIV-1, HCV might also escape the antigen processing and presentation pathways in DCs. 131 HCV bound to blood DCs or hepatic DCs in liver sinusoids may allow the transfer of the virus to the underlying hepatocytes when DCs traverse the sinusoidal lumen in order to gain entry to the hepatic lymph. Beside DCs, liver sinusoidal endothelial cells (LSECs) have also been shown to bind recombinant HCV via interaction of HCV glycoprotein E2 with DC-SIGN and DC-SIGN-related protein (DC-SIGNR or CLEC4M) expressed on the surface of LSECs. 132 However, LSECs have been found to be non-permissive to the entry of HCV pseudovirus, as well as cell culture-derived HCV. 132 Nonetheless, high-affinity binding of HCV to LSEC might still be an important initial step towards its efficient entry into the underlying hepatocytes.
Impact of Chronic HIV-1 and HCV Infection on DCs
Dendritic cells are responsible for orchestrating a successful immune response against viruses; viral persistence might therefore be facilitated by its successful impairment of DC functions. At the primary level, viruses can modulate the frequency of various DC subsets by causing their aberrant trafficking, inducing apoptosis, or interfering with their development. Compared with uninfected individuals, HIV-1-infected60,133–137 as well as HCV-infected138–143 individuals have lower numbers of mDCs and pDCs. During HIV-1 infection, pDCs migrate to the inflamed lymph nodes, where they become activated, apoptotic, and, frequently, infected with the virus.144,145 During HCV infection, blood DCs are enriched in the liver,142,146,147 suggesting that increased migration of DCs to the liver, which is the primary site of HCV replication, causes the observed drop in blood DC count. Other possibilities are that HCV targets DC precursors, as reported by Sansonno et al, 148 or that HCV directly targets DCs to reduce their numbers. In this regard, it has been shown that HCV core, NS3, and NS5 proteins induce apoptosis in mature DCs in vitro. 149 Interestingly, in vitro studies have shown that HCV envelope glycoprotein E2, as well as sera from HCV-infected patients, inhibits the migration of DCs towards CC-chemokine ligand 21 (CCL21), a CCR7 binding chemokine that is important for their homing to lymph nodes. 142 This leads us to an intriguing hypothesis that HCV impairs the ability of DCs to migrate to the draining lymph nodes, causing them to get trapped in the liver. This hypothesis has not been thoroughly investigated in vivo.
Under different immunological states, DCs can either produce IL-12 or IL-10. While IL-12 promotes the differentiation of CD4+ and CD8+ T cells to T 1 cells and CTLs respectively, IL-10 is capable of inhibiting the same. 6 MDDCs, mDCs, and pDCs from HIV-1-infected150, 152 and HCV-infected 141 143 153 154 individuals have been shown to upregulate IL-10 and downregulate IL-12 and IFNα production in response to various maturation stimuli (Fig. 1). Furthermore, the ability of NK cells to mediate early antiviral defense might be affected by dysregulated production of cytokines by DCs. Evidence from several studies indicates that defective pDC function during HIV-1 infection contributes towards impaired NK cell activity, particularly their ability to secrete IFNγ.155,156 Dysregulation of DC-NK cell crosstalk during HIV-1 and HCV infection is discussed in later sections of this study.
Compared to uninfected individuals, DCs isolated from individuals infected with HIV 150 152 and HCV 141 157 159 were found to be less efficient in stimulating T-cell activation and proliferation, as seen in mixed lymphocyte reaction. This inefficient allogenic T-cell stimulation by DCs isolated from HIV-1 99 and HCV-infected 159 patients could be reversed by neutralization of IL-10, suggesting that virus-induced production of IL-10 by DCs could skew the immune response towards tolerance by limiting T-cell activation and proliferation. To gain a different perspective on the impact of HIV-1 and HCV on DCs, various studies have investigated DC function and patients before and after antiviral therapy. Highly active antiretroviral therapy for treating HIV-1 infection has been shown to restore pDC count as well as IFNα production to normal levels. 155 160 Similarly, in HCV-infected patients that were able to clear the virus upon IFN/RBV treatment, the frequency of pDCs increased substantially and ultimately reached levels observed in uninfected controls. 161
Molecular Basis of Virus-DC Interaction
To understand the molecular mechanisms of interaction between these two viruses and DCs, various in vitro studies have used DC subsets isolated from uninfected individuals and exposed them directly to the 162 recombinant infectious virus or viral proteins. Unlike HIV-1 and many other viruses such as influenza virus and herpes simplex virus 1, recombinant and serum-derived HCV have been shown to be poor inducers of IFNα production in pDCs. This suggests that HCV might inhibit IFNα production by pDCs as a strategy to escape IFNα-mediated immune response against itself. In pDCs, TLR7 and TLR9 are endosomal TLRs, responsible for detecting viral RNA and DNA, respectively. Detection of viral RNA or DNA leads to the activation of NF-κB and IFN-regulatory factors, which control the transcription of type I IFNs (IFNα, IFNβ) and various pro-inflammatory cytokines. HCV inhibits IFNα production induced by CpG oligodeoxynucleotide (a TLR9 agonist), but not by resiquimod (a TLR7 agonist).79, 80, 164 HIV-1 is also known to impair IFNα production by pDCs, indicating that impaired production of IFNα by pDCs might be a common strategy employed by many viruses to establish a persistent infection. Hepatitis B virus envelope protein HBsAg and HIV-1 envelope protein gp120 have been shown to impair TLR9- mediated IFNα production through binding to a C-type lectin, blood DC antigen 2 (BDCA2, also known as CLEC4C).162,163 HCV core particles and recombinant noninfectious HCV particles composed of HCV core 79 and envelope glycoproteins E1 and E279,80,164 have been shown to block TLR9-mediated IFNα production, but whether BDCA2 plays a role in that as well is not known. Interestingly, Takahashi et al 165 have provided evidence that pDCs that infiltrate the liver during HCV infection could be a major source of type I IFNs. In these studies, co-cultivation of pDCs with HCV-infected hepatocytes resulted in marked IFN production in proportion to the number of HCV-infected cells. IFN production was mediated by TLR7, and not TLR9, a finding consistent with earlier studies reporting that HCV inhibits TLR9 but not the TLR7-mediated pathway to suppress IFN production.79, 80, 164 This essentially helps explain why infection with HCV leads to a robust expression of IFN-stimulated genes in liver166,167 despite its remarkable ability to attenuate IFN production by infected hepatocytes. 168
Recent studies have shown that persistent viruses can induce the activity of immunosuppressive enzymes in DCs, which leads to suppression of antiviral T-cell response. Tryptophan-catabolizing indoleamine 2,3 dioxygenase (IDO) activity seems to play a central role in the suppressive function of DCs. 169 On direct exposure to HIV-1, DCs induce IDO activity, which leads to inhibition of CD4+ T-cell proliferation in vitro. 170 Additionally, HIV-1-induced IDO activity in pDCs leads to differentiation of naive T cells into CD4+CD25+FOXP3+ regulatory T cells with suppressive function. 171 In simian immunodeficiency virus-infected macaques, IDO activity peaked at the same time as the increase in plasma viremia, as well as expansion of the CD4+CD25+FOXP3+ T-cell subset, which may play an important role in curbing the SIV-specific CD4+ T-cell response. 172 Enhanced IDO activity is found in both HIV-1 and HCV-infected patients; further investigation is thus needed to completely understand its role in the regulation of the cellular immune response against these viruses.
The inability of DCs to facilitate the clearance of HIV-1 and HCV-infection patients leads to chronic immune activation. Systemic chronic immune activation is already believed to be the primary driver of HIV-1 disease progression and the focus is now on understanding its role in the development of liver fibrosis, cirrhosis, or hepatocellular carcinoma in HCV-infected patients. Recent reports have shown that chronic pDC stimulation and IFNα production in HIV-1-infected individuals are associated with higher risk of progression to AIDS. It is thus becoming increasingly important to understand the role of DCs in HIV-1 and HCV-mediated diseases.
Crosstalk between DCs and NK Cells
In addition to their direct antiviral functions, NK cells are capable of regulating antiviral immunity by modulating the function of DCs.173, 179 Crosstalk between DCs and NK cells promotes activation of both cell types. Activated DCs upregulate NK cell effector functions and NK cells, in turn, induce further maturation of DCs. Fernandez et al 173 showed that DCs are able to promote anti-tumor activity of NK cells activity in vivo. Since then, many studies have investigated the role of DCs in the “priming” of NK cells,180,181 a process that involves both cell-cell interactions and cytokine production. Cytokines produced by mDCs, such as IL-12 and IL-18, can promote NK cell production of IFNγ in vitro.175,177,180,181 Moreover, pDC production of type I IFNs and cell-cell contact is essential for promoting NK cell proliferation and cytotoxicity.173,174,176,177 IFNγ produced by activated NK cells promotes TH1 cell polarization.174,178 NK cell-mediated activation of DCs is also known to promote the differentiation of DCs that are more efficient at inducing CTL responses. 179 Furthermore, DC uptake of apoptotic bodies produced as a result of NK cell-mediated lysis of virus-infected cells can promote DC maturation and presentation of viral antigens to T cells. Therefore, in addition to their direct cytotoxic effects, NK cells are capable of boosting the adaptive immune response.
In a process referred to as “DC editing,” NK cells can kill immature DCs. 181 In vitro studies have demonstrated that a low ratio of NK cells to immature DCs promotes DC maturation, whereas a higher ratio of NK cells to immature DCs can result in NK cell-mediated killing of DCs. 179 NKp30 on NK cells plays an important role in this process. 181 It is clear that the crosstalk between DCs and NK cells plays an important role in regulating the antiviral immune response. In the next section we discuss the impact of HIV-1 and HCV on this crosstalk.
HIV-1
Recent evidence has suggested that the crosstalk between DCs and NK cells is disrupted during HIV-1 infection. mDCs from HIV-1-infected individuals show reduced secretion of IL-12, IL-15, and IL-18, resulting in decreased NK cell activation. During HIV-1 infection, activation of NK cells by pDCs is impaired because of their lower responsiveness to type I IFNs. 182 The ability of NK cells to kill immature DCs is reduced in individuals with chronic HIV-1 infection. 183 This defect in NK cell-mediated DC editing appears to be largely the result of an increase in the percentage of CD56– NK cells with impaired NKp30 function. 184 The mechanism of HIV-1-mediated impairment of DC-NK cell crosstalk is not fully understood. The effects of two HIV-1 proteins, Tat and Nef, on DCs and NK cells have been described.185,186 HIV-1-infected DCs have an upregulation of cell death inhibitors that protects them from TRAIL (tumor necrosis factor-related apoptosis-inducing ligand)-mediated NK cell cytotoxicity. This upregulation of cell death inhibitors is mediated by the high-mobility group box 1 protein (HMGB1). 187 Furthermore, the crosstalk of HIV-1- infected DCs with NK cells has been shown to promote viral replication in HIV-1-infected DCs; however, this can be prevented by blocking HMGB1 activity. 188 In addition, increased production of IL-10 during HIV-1 infection limits NK cell-mediated lysis of immature DCs, which results in accumulation of partially mature, poorly immunogenic DCs in the lymph nodes of infected individuals. 189
HCV
Compared to healthy individuals190, 194 and spontaneous resolvers of HCV infection,192, 195, 197 the frequency of NK cells in peripheral blood is reduced in individuals with chronic HCV infection. Additionally, individuals with chronic HCV infection have an increase in NK cell frequency following successful IFN/RBV therapy. IL-15 is a cytokine that plays a pivotal role in NK cell development, proliferation, and function, and therefore reduction in NK cell frequency could be attributed to reduction in IL-15 levels. In this regard, Meier et al have shown a significant reduction in IL-15 levels in HCV patients when compared to healthy controls; they have also shown that exogenous IL-15 is able to rescue the NK cells of HCV-infected patients from apoptosis, resulting in their increased ex vivo proliferation and function. 193 DCs from HCV-infected individuals are deficient in IL-15 production upon stimulation with IFNα 198 and a consequence of this DC dysfunction could be inadequate development and proliferation of NK cells. DCs express MHC class I-related chain A and B (MICA/B), which are ligands for NK cell activating receptor NKG2D. Interestingly, IL-15 is known to induce the expression of MICA/B on DCs; therefore, reduced production of IL-15 during chronic infection with HCV can affect DC activation of NK cells. 198
Human liver epithelial cells express HLA-E, a ligand for CD94/NKG2A expressed on NK cells. In comparison to healthy individuals, NK cells from individuals with chronic HCV infection show higher expression of CD94/NKG2A and produce IL-10 and transforming growth factor-β (TGFβ) when cultured with human liver epithelial cells, most of which express HLA-E. IL-10 and TGFβ production is known to correlate with impaired activation of DCs. A blockade of NKG2A has been demonstrated to restore the ability of NK cells (from HCV-infected individuals) to activate DCs. The blockade also stimulates NK cells to generate TH 1 - polarized CD4+ T cells. It is important to realize, however, that HLA-E may also be expressed on cells other than liver epithelial cells. In fact, HCV is known to stabilize HLA-E expression on intrahepatic APCs; 199 this represents a mechanism by which HCV modulates the NK cell response. The dysregulation of DC-NK cell crosstalk might also have significant consequences for the antiviral T-cell and B-cell responses. The development of therapeutic interventions aimed at enhancing the immune response against HIV-1 and HCV will require a complete understanding of the molecular mechanisms involved in DC-NK cell crosstalk and how it becomes disrupted during chronic HIV-1 and HCV infection.
Modulation of the Immunological Synapse: Role in HIV-1 and HCV Pathogenesis
The generation of specific immune responses against a pathogen requires the interaction between T cells (expressing the cognate receptor) and antigen-loaded mature DCs in the lymph nodes.1,200,201 During this process a specialized cell-cell junction called the immunological synapse (IS) is formed between these two cell types.200–206 Modulation of IS by HIV-1 (Fig. 3) has been identified as one of the important strategies used by HIV-1 to evade the host immune response in order to spread rapidly within the host.
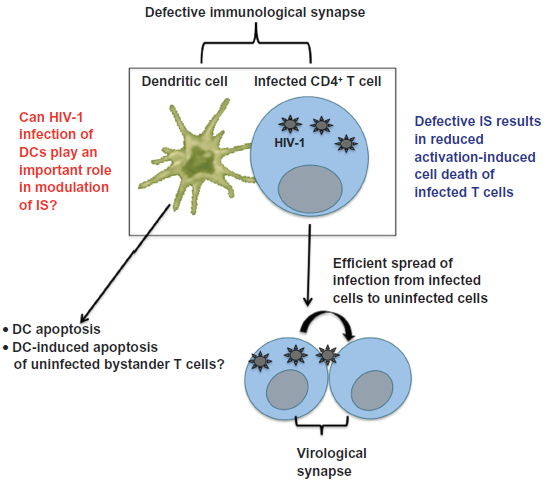
Modulation of immunological synapse by HIV-1 and HCV.
HIV-1
The degree of viral replication in CD4+ T cells depends on their state of activation. In vivo studies have shown that HIV-1 and SIV 172 can replicate in T cells that have a low activation profile, albeit viral replication is much more efficient in activated T cells.207–210 A lack of T-cell activation has been shown to block several steps in the HIV-1 life cycle, including reverse transcription, nuclear import, and integration of the viral genome, as well as transcription and export of multiply spliced viral mRNA after its entry into quiescent CD4+ T cells.90,211–214 Conversely, T-cell activation through T-cell receptor (TCR) ligation or other stimuli often initiates homeostatic programs of apoptosis such as activation-induced cell death (AICD).215,216 The rapid death of infected cells will limit viral production and therefore it is postulated that the virus modulates IS to establish a balance between an apoptosis-prone activation state and the replication-unfavorable environment of resting T cells. 217 HIV-1 protein Nef has been shown to lower the threshold of TCR activation, resulting in induction of an intermediate activation state that prevents AICD and at the same time allows for efficient HIV-1 replication.218–222 This model is also supported by the “bystander effect” hypothesis, which proposes that most of the apoptotic cell death occurring in HIV-1 infection involves uninfected cells rather than infected cells. In addition to facilitating efficient viral replication, infected lymphocytes are also able to efficiently transfer HIV-1 to uninfected lymphocytes through the formation of a virological synapse (Fig. 3). Contrary to what we have discussed above, several groups have shown that various HIV-1 proteins induce apoptosis of infected CD4+ T cells. This could be due to the fact that the actual in vivo concentrations of these proteins seen in HIV-1 infection are largely unknown and in vitro experiments involving overexpression of a particular protein may not be reflective of its in vivo effects. Another interesting point is that many HIV-1 proteins such as Nef and Vpr have been shown to possess both proapoptotic and antiapoptotic effects depending on their expression levels, the activation state of infected T cells, and many other factors.
Several studies have shown that impairment of IS formation between infected lymphocytes and APCs is Nef-dependent.220,223,224 Rabi et al 225 have shown that unstimulated primary CD4+ T cells from HIV-1-positive “elite controllers”—plasma HIV-RNA values persistently below 50 copies/mL throughout the course of infection—are as susceptible to HIV-1 entry and productive infection as viremic patients, suggesting that differences in HIV-1 entry and infection of CD4+ T cells alone cannot explain the elite control of viral replication. It is also known that people who control HIV-1 often have CD4+ T cells that express high amounts of IL-2 and IFNγ in response to HIV peptides. 226 This implies that infected CD4+ T cells can play an important role in controlling the infection and that in addition to ensuring that the HIV-1-infected T cells do not limit viral production by undergoing AICD, impairment of IS by HIV-1 can prevent them from participating in successful clearance of the virus.
Most of the studies about IS have centered on the T-cell side of the IS (IS-T), whereas the functional significance of the DC side (IS-DC) is poorly understood. It is known that DCs from HIV-1-infected patients show increased apoptosis compared to uninfected controls.227, 229 It has been shown that the stable formation of IS results in activation of kinase Akt1. Activation of Akt1 promotes the localization of prosurvival transcription factor NF-κB into the nucleus and proapoptotic transcription factor FOXO1 out of nucleus into the cytoplasm. 230 Therefore, it can be hypothesized that HIV-1-mediated disruption of IS initiates a signaling cascade that results in DC apoptosis. Some surface receptors located at the IS-DC, including ICAM-2, 231 CD22, 232 and CD40, 230 lead to the activation of Akt1 in DCs. Among these receptors, CD40 is particularly interesting as in addition to inducing the activation of Akt1 in DCs, 230 CD40 has been shown to extend their survival. 233 CD40 stimulation results in an increased abundance both of the antiapoptotic Bcl-2 member Bcl-XL and of the cellular inhibitor of apoptosis protein 2 (cIAP-2). 234 This suggests that CD40 might play an important role in the activation of Akt1 and the antiapoptotic signaling relayed from IS-DC. 230 This suggestion is supported by studies in CD40-deficient mice and CD40L-deficient mice that do not elicit CD4+ and CD8+ T-cell-dependent immunity, underscoring the importance of CD40 ligation in prosurvival effects of IS, as well as generation of adaptive immune response. Besides Akt1, Notch has also been implicated in inhibiting the apoptosis of DCs by inducing the activation of STAT3 (signal transducer and activator of transcription 3), a transcription factor known to promote cell survival. Several studies have reported DC-induced apoptosis of T cells.235–240 Because DCs are known to be susceptible to HIV-1 infection, it will be interesting to study whether disruption of IS-DC by HIV-1 is responsible for triggering DC-induced apoptosis of uninfected T cells during IS formation. It will also help to shed light on the role of DCs on bystander T-cell death during HIV-1 infection.
One of the studies has shown that cytoskeletal remodeling mediated by Wiskott-Aldrich syndrome protein (WASp) in DCs is necessary for normal immune synapse formation and T-cell priming. 241 In DCs, the loss-of-function mutation of WASp can also abolish the activation of NK cells by DCs. 242 The HIV-1 pathogenicity factor Nef is known to modulate WASp activity, leading to altered TCR signaling in T cells; 243 in contrast, the effect of HIV-1 on WASp within DCs is not well understood. HIV-1 protein Nef has been shown to trigger the secretion of various cytokines and chemokines (including CC-chemokine ligand 3 [CCL3] and CCL4) by infected APCs.244–247 This can lead to increased HIV-1 replication in CD4+ T cells that get attracted to APCs in response to CCL3 and CCL4 secretion. 244
HCV
As discussed in previous sections of this study, while HCV is primarily a non-lymphotropic virus, it has also been found to replicate in PBMCs as well. Although the impact of HCV on IS has not been investigated thoroughly, we hypothesize that HCV can modulate IS either by infecting DCs and/or T cells or via soluble HCV proteins. Modulation of IS can interfere with T-cell activation, leading to weak T-cell response; additionally, suboptimal activation of T cells can prevent AICD, thus allowing HCV to persist inside them for longer periods. Like HIV-1, HCV could also exploit IS to spread efficiently across PBMCs and this may explain why neutralizing-antibodies against HCV do not always clear the virus.
A widely used approach to assess the antigen-presenting function of DCs is their ability to induce proliferation of allogeneic naïve T cells. Many studies have demonstrated that this function is impaired in MDDCs from patients with HCV infection.153,158 DCs expressing HCV core and E1 proteins have been shown to prime CD4+ T cells abnormally. 248 HCV core protein impairs the priming of specific T-cell responses by DCs even when it is added exogenously in the culture media. 249 This is an important observation because HCV core protein is present in the sera of infected patients and could therefore be responsible for mediating a number of T-cell-inhibitory effects in vitro and in vivo. Krishnadas et al 250 studied the effect of soluble HCV proteins on DCs of healthy uninfected individuals and showed that individual HCV proteins (core, NS3, NS4, NS5) or fused polyprotein (core-NS3-NS4) can impair functions of both immature and mature DCs by downregulating the expression of co-stimulatory and antigen presentation molecules, reducing IL-12 secretion, inducing the expression of Fas ligand (FasL) to mediate apoptosis, interfering with allostimulatory capacity, and inhibiting TLR signaling and nuclear translocation of NF-κB. Interestingly, HCV proteins did not directly inhibit T-cell proliferation, suggesting that they might instead affect the ability of DCs to conjugate with T cells during IS formation. Modulation of IS by both HIV-1 and HCV can have profound effects on the rate of viral replication as well as generation of adaptive immune response. The impairment of IS by these two viruses, especially through their interaction with DCs is gaining a lot of interest. In years to come, we will notice a significant growth in our understanding of IS-DC and how various chronic viruses affect it.
Immune Response in the Liver: Role of Hepatic Antigen Presenting Cells
Besides liver-resident DCs, several other liver cell populations such as LSECs, stellate cells, and Kupffer cells 251 can present antigens and influence the antiviral immune response. However, these APCs exist in a state of active tolerance, thus contributing towards a tolerogenic liver environment, as evidenced by spontaneous acceptance of liver allografts. 252 These liver APCs continuously secrete immunosuppressive cytokines such as IL-10 and TGFβ1. 253 This raises the question of whether the tolerogenic properties of liver APCs contribute towards the establishment of persistent HCV infection and whether liver is a site favoring the evasion of immune response against HCV, as well as other viruses.
Access to liver biopsies from HCV-infected and HIV-1/HCV co-infected patients is limited; isolation of adequate numbers of hepatic APCs is also difficult. Therefore, little is known about the role of hepatic DCs in HCV infection. Kupffer cells and LSECs do not support HCV replication. In vivo studies have demonstrated that hepatic DCs and LSECs can present exogenous antigen to naïve T cells, but this does not lead to effective T-cell response, either due to inhibition of T-cell polyfunctionality or due to acquisition of an immunosuppressive phenotype by T cells. Uptake of viral particles by hepatic APCs may prime CD4+CD25+FOXP3+ regulatory T cells, which are known to dampen the CD8+ T-cell response. This results in failure of the immune system to eradicate the virus from the liver. In chronic HCV infection, antigen-specific CD8+ T cells in liver are frequently reported to become dysfunctional and unable to secrete IFNγ and IL-2. 254 In light of these observations, it is important to advance our understanding of the role of hepatic APCs in priming of HCV-specific T cells.
The antigen presentation ability of hepatocytes has not been studied thoroughly since hepatocytes are generally inaccessible to naive T cells as a result of the barrier formed by LSECs. However, electron microscopy studies have shown that hepatocytes contain microvilli that can pass through fenestrations in the endothelium, allowing them to make contact with the naive T cells in the sinusoidal lumen. 255 Normally, hepatocytes do not express MHC class II molecules, but aberrant expression of MHC class II molecules has been noted in cases of clinical hepatitis.256,257 Therefore, it will not be unreasonable to believe that hepatocytes can induce HCV-specific CD4+ T cells or modulate the antiviral activity of pre-activated CD4+ T cells. Studies on transgenic mice have also shown that hepatocytes can directly stimulate CD8+ T cells, although the stimulated T cells seem to lack cytotoxicity, possibly due to an absence of co-stimulatory molecules on hepatocytes.258,259
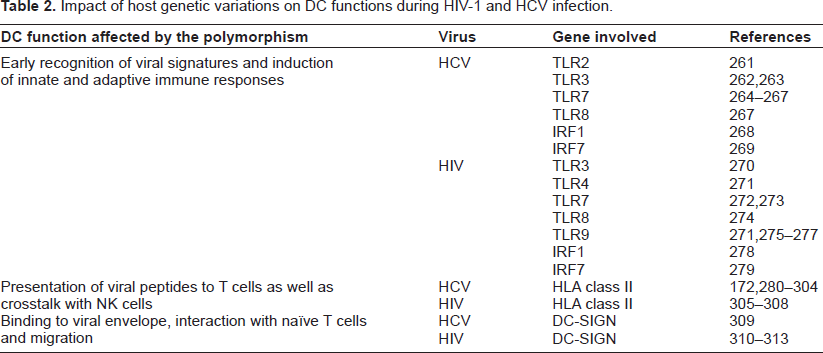
Impact of Host Genetic Variations on DC Functions during HIV-1 and HCV Infection
Studying the role of host genetic variations in determining infection outcome will facilitate our understanding of HIV-1 and HCV pathogenesis. Several genetic polymorphisms have been reported to be associated with low rates of spontaneous clearance, poor treatment response, or accelerated disease progression during the chronic phase of infection. Some of these polymorphisms or rare mutations are found in genes that control DC functions such as recognition of virus, priming of T cells, migration to lymph nodes, and cross talk with NK cells (summarized in Table 2). Understanding the mechanistic underpinnings behind these genetic associations will allow us to gain a deeper insight into the immune evasion strategies adopted by HIV-1 and HCV infection, in addition to helping us devise better treatment options.
Impact of host genetic variations on DC functions during HIV-1 and HCV infection.
Future of Anti-HCV Treatment with and without HIV-1 Co-Infection
Since its introduction in 1996, highly active antiretroviral treatment (HAART) against HIV-1 has been quite successful in controlling HIV-1 infection. The current standard treatment for treating HCV genotype 2–3 infection is a combination of pegylated interferon (PEG-IFN) and an antiviral drug, ribavirin (RBV). Current therapy for HCV genotype 1 consists of an HCV protease inhibitor, telaprevir or boceprevir, in combination with PEG-IFN and RBV. In contrast to HIV-1, treatment of HCV has been difficult because of the low rate of success and a high rate of treatment discontinuation, primarily owing to side effects of IFN/RBV therapy. A sustained virological response is defined as persistent absence of serum HCV RNA for 6 months or longer after therapy and is generally regarded as a cure for HCV infection. However, HCV treatment is toxic and has significant side effects such as anemia, thrombocytopenia, neutropenia, diarrhea, and flulike symptoms such as fever, chills, fatigue, headache, and muscle aches. Therefore, it becomes very important to determine the predictive factors of successful treatment response.
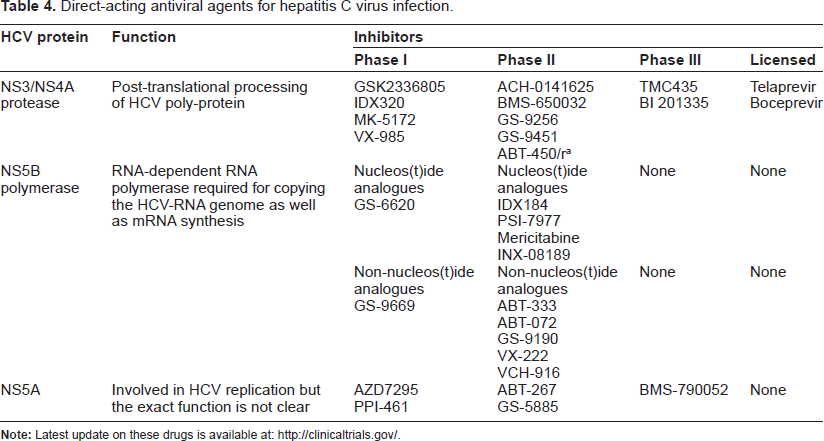
In order to understand the role of DCs in treatment outcome—as well as to study the impact of IFN/RBV therapy on DCs—various studies have attempted to characterize the phenotypic and functional status of blood DCs before or during IFN/RBV therapy. Table 3 highlights those studies that have demonstrated a correlation between the phenotypic and/or functional status of DCs with sustained virological response. Both IFN and RBV have nonspecific and largely unknown mechanisms of action; therefore, various drugs that can directly target HCV at various stages of replication are currently under development (summarized in Table 4). Supplementation of PEG-IFN/RBV with telaprevir or boceprevir in a treatment-naive population improved sustained a virological response of between 40% and 75%. Additionally, treatment duration was shortened from 48 weeks to 24–48 weeks for approximately 50% of patients. In treatment-experienced populations, the treatment response after supplementing PEG-IFN/RBV with telaprevir or boceprevir was ~30% for non-responders, ~55% for partial responders, and 75% to 85% for relapsers. Telaprevir and boceprevir and many other drugs that are in the pipeline will begin a new era of HCV treatment, the ultimate goals of which will be to shorten the duration of therapy, improve sustained virological response rates, and minimize the side effects. Many clinical trials are underway which will address whether these direct-acting antiviral (DAA) drugs will be equally efficacious during HIV-1/HCV co-infection. Moreover, in light of the suggested role of DCs in harboring and disseminating HCV, it will be interesting to study if the efficacy of anti-HCV drugs is dependent on their ability to clear the virus from DCs.
DC-related predictors of sustained virological response to the IFN/RBV therapy.

Weekly administration of PEG-IFNα2b (1.5 μg/kg of body weight) by subcutaneous injection (s.c.) plus oral ribavirin at a dose of 1000–1200 mg, depending on the body weight, for 24 weeks;
PEG-IFNα2b (75–135 μg/week, s.c.) plus oral ribavirin (600–1000 mg/day) for 48 weeks;
PEG-IFNα2b (1.5 μg/kg of body weight/week, s.c.) plus oral ribavirin (800–1200 mg/day) for 6 or 12 months according to the HCV genotype;
PEG-IFNα-2a (180 μg/week, s.c.) plus oral ribavirin (1000–1200 mg/day) for up to 48 weeks;
PEG-IFNα2b (75–135 μg/week, s.c.) plus oral ribavirin (600–1000 mg/day) for 48 weeks.
Direct-acting antiviral agents for hepatitis C virus infection.
Conclusions and Future Perspectives
In the last few years, enough evidence has accumulated to suggest that both HIV-1 and HCV target DCs to cripple the innate and adaptive immune response directed against them. Both viruses affect the ability of mDCs to produce key cytokines that are necessary for the development and activation of an effective T-cell response. In addition, they seem to use similar strategies to attenuate the potent antiviral IFNα response in pDCs. Besides interfering with the natural antiviral activity of DCs, these viruses also use DCs as vehicles for widespread dissemination within the host.
In future, the key issues for improving our understanding of the interaction of HIV-1 and HCV with DCs include the molecular mechanisms of viral entry, processing, and presentation by DCs; the identification of mechanisms that regulate the balance between intrahepatic tolerance and immunity; the impact of HCV, if any, on the formation of immunological synapse between DCs and T cells; molecular mechanisms involved in DC-NK cell cross-talk; and the role of HCV-infected DCs in aiding viral transmission. Answers to these questions will not only help us understand the immunobiology of these two viruses but will also contribute towards the development of better treatment strategies aimed at generating a protective immune response that controls viral infection, without any significant side effects.
Author Contributions
Wrote the first draft of the manuscript: MS, PJ. Contributed to the writing of the manuscript: MS, PJ, ZKK. Agree with manuscript results and conclusions: MS, AHT, ZKK, PJ. Jointly developed the structure and arguments for the paper: MS, PJ. Made critical revisions and approved final version: MS, AHT, ZKK, PJ. All authors reviewed and approved of the final manuscript.
Funding
We wish to acknowledge United States Public Health Service/National Institutes of Health Grants R01 AI077414-01 (to P J) and AI093172 (to Z K) and internal funds from the Department of Microbiology and Immunology, Drexel University College of Medicine to PJ.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.