
Abstract
Objective
To delineate the plasma pharmacokinetics and determine the corneal uptake of valine based stereoisomeric dipeptide prodrugs of acyclovir (ACV) in rats.
Methods
Male Sprague-Dawley rats were used for the study. Pharmacokinetics of ACV, L-valine-acyclovir (LACV), L-valine-D-valine-acyclovir (LDACV) and D-valine-L-valine acyclovir (DLACV) prodrugs were delineated. These compounds were administered intravenously as a bolus via jugular vein cannula and orally by gavage. Samples were purified by protein precipitation method and analyzed by LC-MS/MS. Pertinent pharmacokinetic parameters were obtained by using WinNonlin. Corneal uptake studies of LDACV and LACV were studied following oral administration.
Results
Following
Conclusions
LDACV increased both the oral bioavailability and subsequent
Introduction
Infection with herpes simplex virus (HSV) can be classified as orofacial herpes, genital herpes and herpes keratitis depending on the site of infection. Acyclovir (ACV) is an approved drug of choice for infections caused by both HSV-1 and HSV-2. The current mode of treatment for herpes keratitis includes use of topical trifuorodothymidine (TFT), idoxuridine (IDU) and vidarabine. These agents are highly cytotoxic, thus limiting their usage for long term treatment. Even though, ACV is considered safe, currently, there is no topical formulation of ACV available for the treatment of herpes keratitis in United States. Genital herpes is usually treated with ACV and its analogs/prodrugs administered intravenously or orally. Following initial episode of herpes, chance of recurrence is fairly high due to various factors including fever, trauma, stress, light, immunosuppressive agents and exposure to UV radiation. There is approximately 50% chance of recurrence of herpes keratitis within 2 years of initial episode. 1 Hence continuous suppression rather than intermittent dosing is suggested as the preferred therapeutic intervention.
Systemic drug delivery (intravenous or oral) is potentially an effective route to treat various systemic as well as ocular disorders. However, drugs administered by this route must cross the intestine to reach the systemic circulation and subsequently the blood ocular barriers (BOB) to reach the inner ocular tissues. BOB is comprised of blood aqueous and blood retinal barriers. Transport of hydrophilic molecules like acyclovir and ganciclovir from systemic circulation into anterior chamber tissues like cornea is restricted by blood aqueous barrier (BAB). BAB is a selectively permeable barrier between systemic circulation and ocular aqueous fluids formed by the tight junctions of nonpigmented layer of the ciliary body epithelium and the endothelium of iridial blood vessels. It mostly regulates the inward movement of the compounds from blood into the eye. 2
Limited therapeutic efficacy of ACV against herpes infections following oral administration is due to its poor permeability across oral mucosa. Among various strategies that have been investigated to improve cellular permeability of ACV, prodrugs have been found to be effective and most promising. Transporter targeted prodrug delivery has emerged successfully due to its ability to deliver the drug to targeted tissues and also translocate it intracellularly at a higher rate. 3 7 A wide variety of ester prodrugs of acyclovir were designed to target nutrient transporters including carriers for amino acids and peptides.15,16 Amongst them, peptide transporters (PEPT) are very useful due to their high capacity and wide substrate specificity which provide more flexibility in designing a prodrug. 8 10 Increased oral bioavailability of valacyclovir (VACV), a valine ester prodrug of acyclovir is due its recognition and translocation by peptide transporters present on intestinal mucosa. 11 14 A series of dipeptide esters of ACV were evaluated for corneal and systemic bioavailability following topical and oral administrations.15,16,21
A functionally active PEPT was identified on both the blood aqueous and blood retinal barriers.17,18 PEPT at BAB can be utilized to improve drug delivery of ACV after
Hence enzymatically stable dipeptide conjugates of ACV were synthesized and evaluated for interaction with PEPT, uptake/transport across Caco-2 cells and metabolism in various tissue homogenates. 20 In the same study, the effect of incorporation of a D-isomer upon rate of prodrug hydrolysis was also evaluated. Stereoisomeric dipeptide conjugates had different hydrolytic rates depending on the position of a D-isomer in a dipeptide and also the number of D-isomers. One important observation in that study was that the dipeptide conjugates with one D-isomer did not lose their affinity towards PEPT and also possess enhanced metabolic stability.
Hence, it is important to evaluate the stereoisomeric dipeptide prodrugs
Materials and Methods
Materials
VACV was a gift from GlaxoSmithKline, (Research Triangle Park, NC). L-valine-D-valine-acyclovir-(LDACV) and D-valine-L-valine acyclovir (DLACV) were synthesized in our laboratory according to a standard established procedure. 20 Heparin and heparin coated tubes were obtained from Fisher scientific. Saline was purchased from Sigma chemical Co (St. Louis, MO). All other chemicals were purchased from Sigma chemical Co and were used as received without further purification.
Animals
Jugular vein cannulated male Sprague-Dawley rats weighing between 200 to 250 g were obtained from Charles River Laboratories (Wilmington, MA). Animal care and treatment employed in this research were in compliance with the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the National Institutes of Health.
Pharmacokinetic studies
Prodrugs of ACV equivalent to 20 mg/kg of ACV were administered orally by gavage to rats. VACV, LDACV and DLACV were administered intravenously as a bolus
Corneal uptake studies
For determining the corneal uptake, equimolar doses of prodrugs LACV and LDACV equivalent to 30 mg/kg of ACV were administered to rats by oral gavage. At the time of plasma Tmax (15 min), animals were euthanized with 10 mg per 100 g body weight of sodium pentobarbital. Immediately the eyes balls were proptosed, enucleated carefully and washed with ice-cold with Dulbecco's phosphate buffered saline (DPBS) pH 7.4 to remove any traces of blood and immediately frozen. Subsequently, a small incision was made in the sclera under the magnifying glass and the cornea was carefully excised ensuring that no trace of sclera was present. Corneas were washed with DPBS pH 7.4, dried and weighed.
The cornea was homogenized with a tissue homogenizer (Tissue tearer model 985–370). The homogenate was centrifuged, and the supernatant was collected and frozen at –80 °C. For preparing the calibration standards, blank tissues were collected and treated in a similar manner.
Sample preparation for LC-MS/MS
Samples were purified by protein precipitation method. Ganciclovir (GCV) was used as an internal standard. Plasma and corneal tissue samples were thawed at room temperature. Five μl of 10 μg/ml GCV (500 ng/ml) and 200 μl of acetonitrile were added to 100 μl of sample. The mixture was vortexed vigorously for 2–3 min and centrifuged at 5000 g for 10 min at 4 °C. The supernatant was collected and the solvent was evaporated to dryness with a Speedvac (SAVANT Instruments, Inc., Holbrook, NY). The dry residue was reconstituted in 100 μl water, vortexed for 2 min and further centrifuged (5000 g, 4 min). The obtained supernatant was analyzed using LC-MS/MS. Appropriate calibration standards of ACV and its prodrugs were prepared by spiking a known analyte concentrations to blank plasma and corneal homogenate obtained from untreated rats. The calibration standards were also subjected to identical treatment as samples using a blank corneal matrix and a calibration curve was generated.
LC/MS/MS
A validated LC-MS/MS method for the analysis of dipeptide prodrugs of ACV was established. 22 Briefly, a linear Ion Trap Quadrople LC/MS/MS mass spectrometer (AB Sciex instruments) with electrospray ionization on Turbo Spray ion source (API 2000; Applied Biosystems, Foster City, CA, USA) coupled to an Agilent 1100 binary pump, degasser, and an autosampler was used. Data was collected in multiple reactions monitoring (MRM) mode. Typical ion source parameters were tabulated (Table 1). The entrance potential (EP) was 6 V. The nebulizer gas was set at 10 (arbitrary units), the curtain gas at 8, the collision gas at 4, the ionization voltage at 4500 V and the source temperature was at 500 °C. The compounds were separated with Grace Alltima C8 (150 mm × 2.1 mm, 5 μm pore size) column attached to a guard column. The flow rate was 0.15 ml/min. The prodrugs and their respective metabolites were quantified by the ratio of analyte to the internal standard (ganciclovir).
Mass spectrometric conditions for the LC-MS/MS analysis of ACV and its prodrugs in plasma.

Pharmacokinetic data analysis
All the relevant pharmacokinetic parameters were calculated by noncompartmental analyses of plasma concentration–-time curves with a pharmacokinetic software package WinNonlin, version 5.0 (Pharsight, Mountain View, CA). Cmax, Tmax, Clast and Tlast values were obtained from the log plasma concentration–-time curves. Concentration at zero time point (C0) after
Ct is the last quantitable concentration at time t.
Total body clearance (ClT) and volume of distribution (Vz) after
Results
Pharmacokinetics of LACV and ACV following i.v and oral administrations of LACV
Following

Plasma concentration–-time profiles of LACV, and the regenerated ACV following
Plasma pharmacokinetic parameters of LACV and the regenerated ACV following

Like in

Plasma concentration–-time profiles of LACV, and the generated ACV following oral administration of LACV in rats (♦-LACV, ▪-ACV). Each value is represented as mean ± SD (n = 3–4).
Plasma pharmacokinetic parameters of LACV and the regenerated ACV following oral administration of LACV in rats. (Values are represented as mean ± S.D, n = 3–4).

Pharmacokinetics following i.v and oral administrations of LDACV
Following

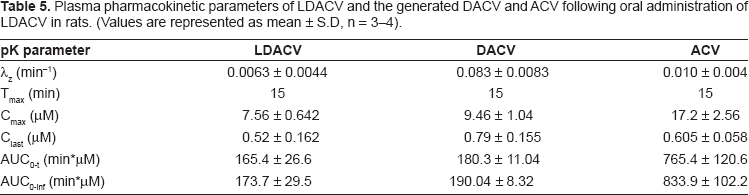
Plasma concentration–-time profiles of LDACV, and the generated DACV and ACV following
Plasma pharmacokinetic parameters of LDACV and the generated DACV and ACV following

Similar to

Plasma concentration–-time profiles of LDACV, the generated DACV and ACV following oral administration of LDACV in rats (♦-DLACV, ▪-ACV). Each value is represented as mean ± SD (n = 3-4).
Plasma pharmacokinetic parameters of LDACV and the generated DACV and ACV following oral administration of LDACV in rats. (Values are represented as mean ± S.D, n = 3–4).
Pharmacokinetics following i.v. and oral administrations of DLACV
Following
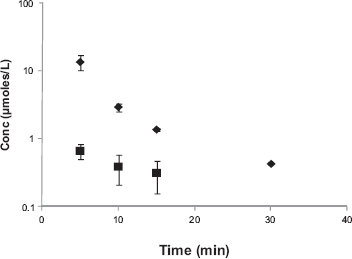
Plasma concentration–-time profiles of DLACV, and the generated ACV following
Plasma pharmacokinetic parameters of DLACV and the generate ACV following
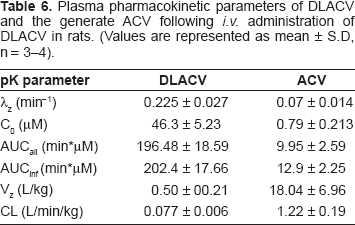
Following oral administration of DLACV, only DLACV and ACV were detected at a very low concentration for a period of 15 and 10 min, respectively. Pharmacokinetic parameters were not calculated since the drug was not detected after the initial few time points. Maximum concentration of DLACV detected in the systemic circulation was 0.53 ± 0.04 μM, which might be due to its poor absorption across intestinal mucosa.
Comparison
Cmax, Tmax and AUC of total drug (prodrug + drug) in the systemic circulation following oral administration of LACV and LDACV were tabulated and compared (Table 7). Based on the Cmax and AUC values, it is evident that both LACV and LDACV have certainly improved the oral bioavailability of ACV. The Cmax and AUC of LDACV are comparable to those of LACV.
Comparison of pharmacokinetic parameters of LACV and LDACV following oral administration to rats. (Each value represents mean ± SD, n = 3–4).

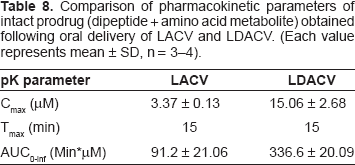
Moreover, the amount of prodrug (dipeptide + amino acid intermediate) observed in the systemic circulation following oral administration of LACV and LDACV were also tabulated and compared (Table 8). Both Cmax and AUC values were in the order of LDACV > LACV. The Cmax and AUC of the intact prodrug resulted following oral administration of LDACV is almost 5 and 4 fold higher than compared to LACV.
Comparison of pharmacokinetic parameters of intact prodrug (dipeptide + amino acid metabolite) obtained following oral delivery of LACV and LDACV. (Each value represents mean ± SD, n = 3–4).
Corneal uptake of ACV following oral administration of LACV and LDACV
The amount of total drug that was detected in cornea following oral administration of LACV and LDACV was compared (Fig. 6). LDACV resulted in almost 2 times higher corneal concentration of ACV compared to LACV. The corneal homogenate was analyzed for all the species. However, only LDACV and ACV were detected in the corneal homogenate with significantly higher concentrations of ACV compared to LDACV in both the eyes (Fig. 7).

Corneal uptake of ACV following oral administration of LDACV and LACV in rats. (Each value represents mean ± SD).

Corneal concentrations of LDACV and ACV following oral administration of LDACV at a molar dose equivalent to 30 mg/kg of acyclovir in rats (□-ACV, ▪-LDACV). Values are represented as mean ± SD, n = 3–4) [R-right eye, L-left eye].
Discussion
The concept of dipeptide prodrugs of acyclovir targeting the peptide transporters on the intestinal mucosa and rabbit cornea has been previously reported from our laboratory.15,16,21 All the dipeptide prodrugs were based on L-amino acid isomers which are natural substrates for hydrolyzing enzymes. However, a recent report from the authors has shown that the incorporation of a D-isomer into a dipeptide can modulate the enzymatic stability of the prodrugs as well as retain the affinity towards PEPT.
20
The effect of rate of prodrug hydrolysis on cellular permeability was also studied in the previous report using valine based stereoisomeric dipeptide prodrugs of acyclovir. In this research, the stereoisomeric dipeptide prodrug concept was extended to
Following
Oral administration of LACV resulted in both the prodrug and drug. However, Cmax and AUC of ACV were significantly higher than LACV (Table 3). This rapid and complete hydrolysis of LACV is in agreement with published literature. Unlike the results obtained after
Cmax and AUC values observed after oral delivery of LACV and LDACV are tabulated and compared (Table 7). Both LACV and LDACV have enhanced the oral bioavailability of ACV by 5–6 folds. The Cmax and AUC values observed after oral delivery of LDACV were comparable to that of LACV. However, the amount of intact prodrug (dipeptide + amino acid metabolite) resulting with LDACV was almost 4–5 fold higher than of LACV (Table 8). Thus, LDACV has not only improved the oral bioavailability of ACV but also resulted in significant concentrations of intact prodrug. Since the ultimate objective is to evaluate whether the resulted intact prodrug in the systemic circulation would aid in enhancing the corneal absorption of ACV, corneal concentrations were analyzed for total drug following oral dosing of LDACV LACV was used as a control. The concentrations in the cornea following LDACV was 2 fold higher than those resulted after administration of LACV (Fig. 6). Furthermore, in the cornea, LDACV has resulted in both LDACV and ACV. However the concentrations of ACV were much higher than LDACV in both the eyes indicating the hydrolytic capability of ocular tissues (Fig. 7). The higher corneal concentrations of ACV observed after LDACV might be due to the recognition and translocation of the prodrug from the systemic circulation into cornea by PEPT expressed on the blood aqueous barrier (BAB) followed by hydrolysis by proteases and/or esterases in iris-ciliary body, aqueous humor and cornea. The ability of ocular tissues to hydrolyze these prodrugs was reported.15,16,21 This clearly indicates that the intact prodrug concentrations in the systemic circulation would be helpful to improve the concentrations of the drugs in cornea by targeting the transporters at BAB.
In summary, of all the stereoisomeric dipeptide prodrugs evaluated, LDACV improved both the systemic and corneal absorption of acyclovir after oral administration. Thus it is a promising candidate for further preclinical and clinical studies for the treatment of both genital herpes and corneal keratitis. Pharmacokinetics of stereoisomeric dipeptide prodrugs provided significant insight into their metabolism and absorption across intestinal mucosa and BAB. This information will aid in designing various other dipeptide promoieties of other therapeutic molecules. This concept can be further extended to deliver drugs to the posterior segment of the eye following oral administration.
Disclosures
The authors report no conflicts of interest.
Footnotes
Acknowledgements
The authors would like to acknowledge Dr. Swapan K. Samanta for synthesizing the prodrugs. We would like to thank GlaxoSmithKline for the generous supply of Valacyclovir. This work was supported by NIH grants RO1 EY09171-14 and RO1 EY10659-12.