
Abstract
Proteinase-K resistant prion protein (PrPres) has the property to aggregate in TSE-injured animal tissues. We have developed a test method to discriminate scrapie-infected and mock-infected hamsters by detecting the PrPres in plasma. It seemed that aggregation of the PrPres with some heterogeneous molecule(s) enabled successful detection by this method. In order to investigate which molecule became the partner in the PrPres aggregates; we examined some molecules that could presumably have this ability. As a result, we found synthetic Poly-A RNA, especially in its denatured form, to be the most effective entity although glycoprotein, sulfated polysaccharide showed less effectiveness. DNA in the denatured form also has a high affinity, although in the presence of protein the effectiveness unsuccessful. On the basis of this result, it is possible that the PrPres aggregate in scrapie-infected hamster plasma is composed of PrPres and RNA.
Keywords
Introduction
Transmissible spongiform encephalopathy (TSE) is a fatal neurodegenerative disease, which is represented as variant Creutzfeldt-Jakob disease (vCJD) and the so-called classical CJD in humans, bovine spongiform encephalopathy (BSE) in bovines, and scrapie in sheep and goats. The pathological manifestations of these diseases are characterized by sponge-like pathological appearance and deposition of amyloid plaques composed of disease-related PK-resistant prion proteins (PrPres) around the affected area. Hence, the diagnosis of these diseases is generally confirmed on the basis of the pathological observation of the affected tissue and biochemical detection of the PrPres in the postmortem test. 1 In order to enhance survival rates in such patients, it was considered desirable to develop an antemortem test that could enable the diagnosis of the disease as early as possible. Thus, a pathological test using biopsy-derived tonsil or appendix tissue was proposed.2,3 Since the sensitivity of the tonsil- and appendix-based test was limited, the development of a more convenient method using blood samples (i.e. a blood test) was suggested. In the UK, 4 cases of transfusion-related TSE transmission, and a case of infection in a hemophiliac patient who used a clotting factor continuously were recently discovered. These findings suggest that transfusion or use of plasma products caused the TSE infection which raised great concern regarding the iatrogenic secondary transmission of this disease.4–6 Hence, a pre-symptomatic test rather than an antemortem test was more desirable, not only to treat patients but also to identify infected individuals from healthy looking persons to prevent horizontal transmission of TSE via blood. For conducting epidemiological studies, a pathological test using the biopsy-derived tonsil and appendix tissues was previously carried out.7–9 Since the tonsil- and appendix-based tests only yielded primary data regarding the number of healthy carriers in the general population, it became desirable to develop a blood test immediately to confirm TSE in the general population; thus, the development of a blood test became one of the primary objectives of TSE research. 10 Transmission of TSE via blood was also observed in transfusion experiments performed using sheep that were infected with scrapie, indicating that not only vCJD but also other form of TSE could be transmitted via blood.10,11
The infectivity titer of TSE in the blood was found to be 1–2 log10 LD50/ml, which was extremely low compared to that found in the brains of infected animals. 10 This indicates that only a minimum amount of the PrPres is present in the blood. In addition, large amounts of blood proteins prevent the detection of the PrPres in the blood, thereby rendering the development of a blood test more difficult. It is commonly assumed that PrPres exist as aggregates in the blood. Whether such aggregates or the small amyloid particles are the suspected infectious agents of TSE is yet unknown. Despite this uncertainty, it is assumed that the success or failure of a blood test may depend on that of the detection of PrPres micro aggregates in the blood.12,13
Several substances are known to display affinity to PrPres. Of such substances, glycoproteins (plasmin and plasmionogen), sulfated polysaccharides (heparan sulfate), and free polynucleotide (DNA and RNA) in the fragmented form are known to exist in the blood plasma.14–21 It has also been reported that the presence of a polyanionic molecule, especially Poly A, is necessary for the in vitro multiplication of PrPres, as detected by protein misfolding cyclic amplification (PMCA).22–25
We previously attempted to develop a blood test system using the plasma of scrapie-infected hamsters. 26 This system was designated as “acidic sodium dodecyl sulfate (SDS) precipitation (abbreviated to ASP).” This system was able to effectively discriminate between infected and mock-infected hamsters by analyzing blood samples. The experimental data obtained using the ASP system suggested that the PrPres could be isolated in the form of aggregates of heterogeneous molecules. Since the ASP method can easily and rapidly detect the PrPres in the plasma from infected animals, we hypothesized that this method could be used as a basis for the development of an efficient blood test system. In order to make the blood test more efficient, it is necessary to determine the partner molecule(s) in the PrPres aggregates. In addition, determination of aggregation partners is expected to establish the nature of the aggregates that contain PrPres. In this study, using the brain homogenates of scrapie-infected hamsters, we showed that denatured nucleic acids (both DNA and RNA) could become aggregation partner molecules with PrPres and form firm precipitates after ASP. However, the effectiveness level of poly nucleotides in the aggregates varied depending on the weakly sequenced specific manner and differences of RNA or DNA. Presence or absence of peptide-N-glycosidase F (PNGase F) protein also affected the ability of poly nucleotides to acquire enhanced ability of aggregation with the PrPres.
Materials and Methods
Antibody and enzymes
Anti-PrP antibody, 3F4 (Signet) monoclonal antibody (mAb) and horse-radish peroxidase-conjugated goat anti-mouse IgG (HRPGAM) which was a composite of the chemiluminescence substrate kit (Thermo Co. Ltd), were used as the primary and secondary antibodies for the detection of PrP molecules in immunoblot. Proteinase K (PK) was purchased from Merck Co. (specific activity, 40 m Anson U/mg protein). PNGaseF was purchased from Roche Diagnostics Co. Ltd. (specific activity, 25,000 U/mg protein).
Glycoproteins, sulfated polysaccharides, and polynucleotides
Plasmine (Plsm) and Plasminogen (Plsmg) derived from human plasma were purchased from Sigma Co. Heparan Sulfate (HepSul) derived from pork intestine and fetuin (Fet) derived from fetal bovine plasma were both purchased also from Sigma Co. Synthetic Poly A (Poly A; 20 mer) and Poly C (Poly C; 20 mer) were synthesized by Quiagen Co. Ltd. Sheared salmon sperm DNA (SSpDNA) was purchased from Ambion Co. Blood of a mock infected hamster was collected from healthy-hamster brain homogenate-inoculated hamsters on day 54 after inoculation. This blood was immediately centrifuged at 2,000 × g 10 min to separate the cellular fraction and the plasma; the plasma was then collected as the mock-infected hamster plasma (mcPl) thereafter. All the above mentioned reagents were used as the mixing partner molecules of PrPres aggregates to examine their associability.
Scrapie-infected and mock infected hamster brain homogenates (scBrh, mcBrh)
Scrapie (sc237 strain)-infected hamsters were kindly provided by Drs. Yokoyama and Takata (Research Center for Prion Diseases, National Institute of Animal Health, Japan). The brain homogenates of terminal disease stage sc237-infected hamsters was intracerebrally inoculated into healthy male hamsters (8-weeks-old); the inoculated hamsters were killed 54 days after inoculation and disease progression was confirmed. Brain homogenates from a healthy hamster of the same age as that of the infected animal was inoculated intracerebrally into another healthy male hamster as mock infection; the inoculated hamsters were killed at 54 days after inoculation. Brains from the sc237 infected (scBrh) and mock-infected (mcBrh) hamsters were homogenized in 25 mM Tris buffered saline (TBS; pH 7.2) containing 0.5% triton × 100, 0.5% deoxycholate, and a protease inhibitor cocktail (Sigma) into 10% homogenates. These homogenates were centrifuged at 2,000 × g 10 min, and the sediment was discarded.
Enzyme treatment
The PK treatment of the scBrh and mcBrh was carried out as follows; 2-fold diluted form of each sc- or mcBrh by TBS was mixed with equal volumes of PK solution (final 50 μg/ml) in TBS and incubated at 37 °C for 60 min. The enzymatic reaction was halted by adding 1 mM pefablock, and the resulting mixture was mixed with SDS (finally 3%), DTT (finally 50 mM) and EDTA (2 mM), final concentrations of this as indicated in the parenthesis, was then boiled for 10 min. The resulting preparations were stored in aliquots at -80 °C until use.
Deglycosidation of the suspected glycoprotein partner molecules that had to be tested was performed according to the manufacturer's instruction manual. In brief, the reaction mixture contained 50 μl of 10 μg/ml substances and 3.3 units PNGase F in the 200 μl of deglycosidation buffer. The composition of the deglycosidation buffer was 100 mM phosphate buffered saline containing 1% NP40, 0.1% SDS, 20 mM EDTA, and 1% 2-mercapto ethanol. The reaction mixture was then incubated overnight at 37 °C. The resultant deglycosylated substances were precipitated using 4-fold volumes of methanol, and the precipitates were dissolved using TBS containing 3% SDS and 2 mM EDTA (TBS-SDS) to a concentration of 10 μg/ml then stored in aliquot at -20 °C. HepSul and the three polynucleotides were also subjected or remained untreated with the deglycosidation buffer similarly to examine if pretreatment has any effect on the nonglycoprotein substances for the final ASP precipitates.
Preparation of denatured polynucleotide
One hundred micro liters of Poly A, Poly C, and SSpDNA (10 μg/ml) were separately inoculated into 300 μl of the nucleotide denaturing buffer which was composed of 100 mM phosphate buffered saline containing 20 mM EDTA and 1% 2-mercaptoethanol. These mixtures were incubated overnight at 37 °C. The denatured polynucleotide was then precipitated via ethanol precipitation, redissolved in TBS-SDS to a final concentration of 10 μg/ml, and stored at -20 °C. No additional proteins were included in the denaturing reactions of these polynucleotides. The initials A, C, and D denotes the Poly A, Poly C and SSpDNA in the Figures, respectively.
Test of associability between the suspected partner substances and the PrPres in PK-scBrh
Stored PK-scBrh and PK-mcBrh were dissolved at room temperature. In the first experiment (Fig. 1), 1 μl of the PK-scBrh or the PK-mcBrh were used to indicate the positive and negative controls for PrPres without ASP. In later experiments, 10 μl (Figs. 2 and 3) or 5 μl (Figs. 4 and 5) of their 10-fold diluted preparations were used for the positive and negative controls without subjecting the ASP. For examining the associability between PrPres and suspected partner substances, 5 μl of none diluted PK-scBrh or PK-mcBrh were mixed with 10 μl of 10 μg/ml concentrate of mixing partner substances in the first experiment (Fig. 1) whereas 2 μl of non-diluted PK-scBrh or PK-mcBrh were mixed with 10 μl of 10 μg/ml concentrate of partner molecules in the later experiments (Figs. 2–5) and incubated at 37 °C for 30 min. After incubation, the reaction mixtures were immediately chilled on ice and allowed to stand for a further 10 min on ice. The chilled mixtures were returned to room temperature and, subsequently, subjected to the ASP method.
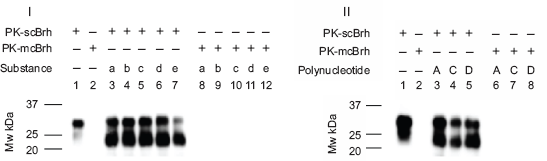
Affinity association of glycoproteins, heparan sulfate, and polynucleotide. Substances of 5 non-nucleotides and 3 poly nucleotide were tested to determine whether they showed associability to PrPres. In panel I, lanes 1 and 2 indicated the immunoblot patterns of scBrh and mcBrh in which 1 μl of non-diluted PK-scBrh and PK-mcBrh were run on the electrophoresis without subjecting them to ASP in order to have a positive and negative control of the PrPres. For examining the associability (lanes 3–12), 5 μl of PK-scBrh or PK-mcBrh were mixed with 10 μl of 10 μg/ml substances and subjected to the ASP thereafter and analyzed by chemiluminescence immunoblot method finally. In this experiment, substances indicated below were used as the mixing partners with PK-scBrh: lane 3; Plsm (a), 4; Plsmg (b), 5; Fet (c), 6; HepSul (d), and 7; mcPl (e). Lanes 8–12 indicate the immunoblot pattern using the substances of a–e as mixing partners of PK-mcBrh. In Panel II, immunoblot patterns of ASP precipitates using 3 polynucleotides as mixing partners of PK-scBrh and PK-mcBrh were indicated. In this panel, lanes 1 and 2 were the positive and negative controls as preprocessed similarly as the panel I. Lanes 3–5 indicated the immunoblot patterns of the ASP precipitates using following substances as mixing partners of the PK-scBrh: lane 3; Poly A RNA (A), lane 4; Poly C RNA (C), and lane 5; SSpDNA (D). Lanes 6–8 indicate the immunoblot pattern of the ASP precipitates using the same polynucleotides and the PK-mcBrh: lane 6; Poly A RNA (A), lane 7; Poly C RNA (C), and lane 8; SSpDNA (D).
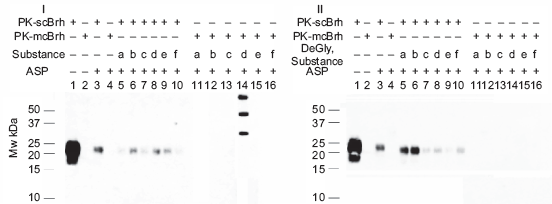
Effect of deglycosylation of the non-nucleotide substances on their affinity association to the PK-resistant PrP (PrPres). The 6 non-nucleotide substances were first treated with the PNGase F or remained untreated, then the effect of this PNGase F treatment on the affinity association between these substances and PrPres in PK-scBrh or PK-mcBrh was tested. Panels I and II indicate the immunoblot pattern of the ASP precipitates between the non-nucleotide substances before (Panel I) and after deglycosylated (Panel II) and PK-scBrh or PK-mcBrh. Processes used in these experiments is described in the Materials and Methods. In both panels, lanes 5(a)–10(f) indicates the results for using following non-nucleotide mixing partners of the PK-scBrh whereas 11–16 indicates the results for using the same non-nucleotide mixing partners of the PK-mcBrh. In these, mixing partners are 5; Plsm (a), 6; Plsmg (b), 7; Fet (c), 8; HepSul (d), 9; mcPl (e) and 10; BSA (f), 11; Plsm (a), 12; Plsmg (b), 13; Fet (c), 14; HepSul (d), 15; mcPl (e), 16; BSA (f). Lanes 1 and 3 show the pattern for PK-scBrh and 2 and 4 show the pattern for PK-mcBrh; lanes 1 and 2 are unprocessed 10 μl of 10-fold diluted PK-scBrh- and PK-mcBrh, while lanes 3 and 4 show the pattern of 2 μl of non-diluted PK-scBrh and PK-mcBrh after ASP, respectively.
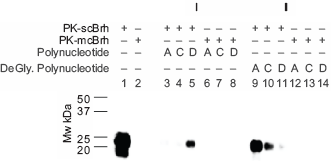
Effect of deglycosylation conditions on the affinity association of the 3 polynucleotides to the PrPres in scBrh. The 3 polynucleotides were incubated overnight at 37 °C in the deglycosylation buffer or remained untreated. Next, 10 μl of the above treated or untreated polynucleotides were mixed with 2 μl of PK-scBrh or PK-mcBrh and subjected to the ASP as described in the materials and methods. In this Figure, Panel I shows the immunoblot pattern of the ASP precipitates obtained from the mixture of untreated polynucleotides and scBrh or mcBrh. Panel II shows the immunoblot pattern of the ASP precipitates from the treated polynucleotide and the PK-scBrh or PK-mcBrh. The following polynucleotide was tested: A, Poly A RNA; C, Poly C RNA; and D; SSpDNA. In both panels, lanes 1 and 2 indicate the immunoblot patterns of the 10 μl of 10-fold diluted PK-scBrh and PK-mcBrh in the absence of any polynucleotides. In Panel I, lanes 3–5 are the immunoblot patterns of the ASP precipitates from the mixture of PK-scBrh and the untreated polynucleotide, lanes 6–8 are the immunoblot patterns of the ASP precipitates from the mixture of PK-mcBrh and the untreated polynucleotide. In panel II, lanes 9–11 are the immunoblot patterns of PrPres in the ASP precipitates from the mixture of deglycosylation buffer-treated polynucleotide and PK-scBrh, lanes 12–14, immunoblot patterns of the ASP precipitates from the mixture of deglycosylation buffer-treated polynucleotide and PK-mcBrh.
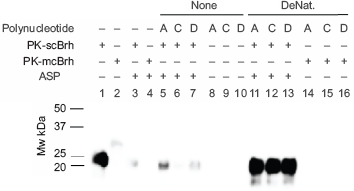
Effect of denaturing on the associability of the 3 polynucleotide to the PrPres. The 3 polynucleotide were denatured or remained non-denatured as described in the Materials and Methods section. The denatured or non-denatured polynucleotide (10 μl) was mixed with 2 μl of PK-scBrh or PK-mcBrh, and examined the effect of denaturalizing to PrPres recovery in the ASP precipitates. The following polynucleotides are shown in the Figure: A; Poly A, C; Poly C, and D; SSpDNA. Tests using the 3 polynucleotides before and after denaturing are indicated as None and DeNat in this Figure, respectively. Lane 1; positive control for PrPres in PK-scBrh, 2; negative control for PrPres in PK-mcBrh, 3; control for the ASP of PK-scBrh, 4; negative control for the ASP using PK-mcBrh, and 5–7; ASP precipitates from mixtures of non-denaturalized polynucleotides and PK-scBrh, in which: lane 5; Poly A, lane 6; Poly C, and lane 7; SSpDNA. Lanes 8–10 indicate the ASP precipitates from mixtures of non-denatured polynucleotide and PK-mcBrh in which used polynucleotide are: lane 8; Poly A, lane 9; Poly C, and lane 10; SSpDNA. Lanes 11–13: ASP precipitates from the mixture of the denatured nucleotides and PK-scBrh in which: lane 11; denatured Poly A, lane 12; denatured Poly C, and lane 13; denatured SSpDNA. Lanes 14–16; ASP precipitates from the mixture of the denatured polynucleotides and PK-mcBrh in which lane 14; denatured Poly A, lane 15; denatured Poly C, lane 16; denatured SSpDNA.
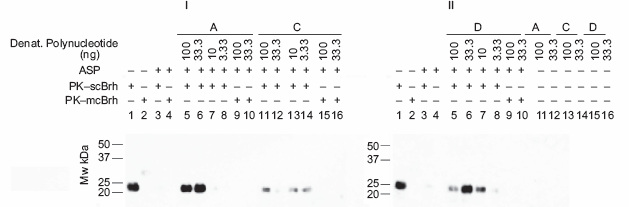
Efficacies of the 3 denatured polynucleotide for aggregating with the PrPres in PK-scBrh. The efficacies of the 3 denatured poly nucleotides in association of the PrPres with PK-scBrh were tested. The denatured poly nucleotides were serially diluted to 3-fold manner and were mixed with PK-scBrh or PK-mcBrh, incubated, and subjected to ASP. The amount of PrPres in the final ASP precipitates were analyzed by the chemiluminescence detection method. In panel I, Lanes 1–4 indicate the control experiments in which lanes 1 and 2 are PK-scBrh and PK-mcBrh without processing the ASP, respectively. Lanes 3 and 4 are PK-scBrh and PK-mcBrh processed with the ASP method, respectively. Lanes 5–8 show the efficacy of PrPres detection by using serially diluted denatured Poly A mixed with PK-scBrh. Lanes 9 and 10; diluted preparation of denatured Poly A was mixed with PK-mcBrh. Similarly, lanes 11–14; serial dilution of the denatured Poly C and PK-scBrh mixture, lanes 15 and 16; serial dilution of the denatured Poly C and PK-mcBrh mixture. In the panel II, lanes 1–4; positive and negative controls in which lanes 1 and 2 indicate PK-scBrh and PK-mcBrh without processing the ASP and lanes 3 and 4; indicate PK-scBrh and PK-mcBrh processed to the ASP. Lanes 5–8; serial dilution of denatured SSpDNA was mixed with the PK-scBrh. Lanes 9 and 10; serial dilution of denatured SSpDNA was mixed with PK-mcBrh. The absence of nonspecific staining in the 3 denatured polynucleotide is shown in lanes 11 and 12 for denaturized Poly A; lanes 13 and 14 for denatured poly C; and lanes 15 and 16 for SSpDNA. In the both panels, initials A, C, and D mean Poly A, Poly C, and SSpDNA, respectively.
Acidic SDS precipitation (ASP)
In brief, PK-scBrh or PK-mcBrh and each of the reaction mixtures above mentioned were mixed with equal volumes of 2 × acidic saline (0.3 M NaCl containing 0.02 M acetic acid and 2 mM EDTA), incubated for 5 min at 10 °C, and centrifuged at 20,000 g for 10 min at 10 °C. Precipitates were collected and dissolved into their original volumes by TBS-SDS, mixed with equal amount of the acidic saline and incubated again. After the incubation the mixtures underwent centrifugation again at 20,000 × g, the precipitates were collected and rinsed with 200 μl of cold methanol then dissolved finally in 15 μl of 2 × SDS sample buffer.
SDS-PAGE and WB
The above mentioned preparations were heated at 100 °C for 10 min and electrophoresis was performed by Laemli's conditions using 15% poly acrylamide gel. After the electrophoresis, the separated proteins were blotted onto poly vinylidene fluoride (PVDF) membrane under the transfer conditions of 50 V and 100 mA for 240 min. In addition non specific sites of the blotted membrane were blocked using Super Block (Thermo Co) for 1 h at room temperature or further overnight at 4 °C.
PrPres signal detection via ultrasensitive chemiluminescence immunoblot method
Blotted and blocked PVDF membranes were rinsed 3 times in TBST (TBS containing 0.05% Tween20). Primary mAb was dissolved in the super block solution containing 0.01% bovine serum albumin (BSA) and 10% Block Ace (Dai Nippon Pharm. Co. Ltd.) and poured onto the blotted and rinsed membrane. The membrane was then gently shaken on a reciprocal shaker overnight at 4 °C. Subsequently, the membrane was washed 5 times with TBST (10 min per a wash). After washing, HRPGAM was dissolved in the Super block solution containing 0.1% BSA and 10% Block Ace was poured onto the washed membrane that was gently shaken for 1–1.5 h on a rotary shaker at room temperature. After incubation with the 2nd Ab, the membrane was again washed for 5 more times with the TBST. The chemiluminescence substrate (Super Signal West Femto Maximum sensitivity substrate; Thermo Co.) was finally poured over the antibody-reacted membrane, and the membrane was left in the substrate for 5 min at room temperature. Chemiluminescence signals corresponding to the primary-antibody-reacted protein bands were detected using an image analyzer LAS 3000 (Fuji film Co. Ltd.).
Results
ASP of scBrh and suspected partner substance mixtures
PK-scBrh or PK-mcBrh and the heterogeneous substances that were suspected to have a binding affinity to PrPres were mixed and subjected to the ASP method, if such substances indicated some affinity in the precipitation reaction (Fig. 1). The following substances were tested: Plsm, Plsmg, Fet, HepSul, mcPl, Poly A, Poly C, and SSpDNA.
Panel I of Figure 1 shows the amount of PrPres detected in the ASP precipitates from the mixtures of PK-scBrh or PK-mcBrh and the above mentioned 5 non-nucleotide substances, while Panel II shows the amount of PrPres detected in the ASP precipitates from the mixture of PK-scBrh or PK-mcBrh and the 3 polynucleotides. In this study, 5 μl of PK-scBrh or PK-mcBrh and 10 μl in 10 μg/ml concentrates of the above mentioned substances were mixed together as described in the Materials and Methods. These mixtures were then subjected to the ASP. One micro liter of PK-scBrh and PK-mcBrh were not processed to the ASP without mixing the suspected binding molecules and used as positive and negative controls of the PrPres, respectively (both panel I and II). Using this procedure, we could determine whether the ASP method indicated the associability between these substances and the PrPres in PK-scBrh. The PK-scBrh without ASP showed a dense band at approximately 30 kDa and a weak band at approximately 23–25 kDa. PK-mcBrh showed a slight band (lanes 1 and 2, respectively). In contrast, the 23–25-kDa bands were denser than the 30-kDa band when PK-scBrh was mixed with the non-nucleotide substances and processed for ASP (Fig. 1, Panel I). When each of the 3 nucleotides were mixed with PK-scBrh, similar banding patterns although slightly weak for the Poly C and PK-scBrh-mixture were observed (Fig. 1, Panel II). No such bands were observed when PK-mcBrh and 5 nonnucleotide or the 3 poly nucleotide mixtures were subjected to ASP (Fig. 1, Panels I and II). Since these banding patterns suggested the presumable affinity of these substances to the PrPres in scBrh after ASP, these substances were suspected as the presumable partner substances of PrPres aggregates. This pattern also suggested that these substances had high affinities to PrPres in scBrh, even though scBrh was preheated in the presence of SDS.
Effect of deglycosidation on glycoprotein partner substances
Of the non-nucleotide substances, since 3 glycoproteins and mock-infected hamster plasma (glycoprotein mixture) were found to have an affinity to PrPres, we investigated whether the saccharide chains on these substances played any role in enhancing their binding affinity. First, these substances were deglycosylated and then mixed with PK-scBrh or mcBrh. These mixtures were subjected to the ASP method and whether PrPres were recovered in the final precipitates was examined (Fig. 2, Panels I and II). As indicated in Panel I, relatively small amounts of PrPres were recovered in the precipitates of PK-scBrh when the suspected partner substances were not mixed. Deglycosylation of these substances did not dramatically affect PrPres recovery rather that it is deglycosylated Plasm and Plsmg that yielded greater amounts of PrPres recovery than the glycosylated ones. mcPl indicated a slight decline in PrPres recovery, and Fet did not affect the recovery process. Treatment of HepSul with the deglycosidation buffer did not affect the binding at all. When these substances were mixed with PK-mcBrh, no PrPres recovery occurred. Although the mixture of the mcPl and PK-mcBrh yielded 3 bands of high molecular weight, the intensity of these bands drastically reduced after deglycosylation of PK-mcPl (lane 15, Panels I and II).
Affinity of the polynucleotide for precipitation
The affinity association of Poly A, Poly C, and SSpDNA to PrPres in scBrh were analyzed (Figs. 3 and 4). First, these 3 polynucleotides were treated or remained untreated in the deglycosylation buffer, which included PNGase F and NP40, and mixed with PK-scBrh or PK-mcBrh, thereafter. These mixtures were then subjected to the ASP and the amounts of PrPres obtained in the precipitates was determined (Fig. 3). After untreated preparations of the 3 poly nucleotides were mixed with PK-scBrh, effective recovery of the 23-kDa PrPres band was observed in only the precipitates from the mixture of SSpDNA and PK-scBrh. On the other hand, mixing of deglycosidation buffer-treated polynucleotides with scBrh did not result in the uniform effectiveness on the PrPres recovery; the most effective recovery was observed for the precipitates from Poly A mixture, followed from the Poly C mixture and the lowest was shown from SSpDNA mixture. These results suggested that the recovery of PrPres in the ASP precipitates was affected by treating the poly nucleotides in the deglycosidation buffer in the presence of denaturing substances. Hence, the 3 polynucleotides were treated first in the nucleotide denaturing buffer, which did not contain NP40 and PNGase F protein (Fig. 4) and the affinity association of these denatured polynucleotides to PrPres in PK-scBrh was examined after mixing and ASP processing. When the recoveries of the PrPres in the precipitates from the mixtures after such processing was examined, enormous amounts of the 23–25-kDa PrPres proteins were obtained in all 3 denatured polynucleotide mixtures (Fig. 4). When ASP of the PK-scBrh was performed without any other nucleotides mixing, only a small amount of the 23-kDa PrPres band was obtained in the precipitates. Therefore, it was clearly indicated that denaturing of the 3 polynucleotides resulted in acquisition in their ability to associate with PrPres to precipitate after ASP (Fig. 4). The 3 polynucleotides did not precipitate by themselves after ASP and did not react with the primary antibody irrespective of their denaturation status. Furthermore, mixing of the both none-denatured or denatured polynucleotide with PK-mcBrh did not yield the 23-kDa PrPres protein band (Fig. 4, Panels I and II). Therefore, this precipitation phenomenon in association with the poly nucleotides was specific for scrapie infection.
Comparison of the association effectiveness between the nucleotides
The association effectiveness of the 3 denaturalized nucleotides to the PrPres in scBrh was compared. The 3 polynucleotides were denatured first, as mentioned in the Materials and Methods, then diluted in TBS-SDS by serially 3-fold manner, and mixed with PK-scBrh or PK-mcBrh thereafter. The mixed preparations were subjected to the ASP and the amount of PrPres in the final precipitates was determined. In Figure 5, the effect of dilution of the 3 denatured nucleotides on the detection of PrPres in the final precipitates was indicated. For 10 μl of 2% scBrh, 33.3 ng of Poly A or SSpDNA was the most effective amount to achieve the highest recoveries to precipitate the 23-kDa PrPres. In contrast, denatured Poly C did not yield such clear results. Therefore, after denaturing, Poly A and SSpDNA were shown to have equal effectiveness at the mixing ratio of 2 μl scBrh: 33.3 ng Poly A or SSpDNA on the affinity association to precipitate the 23-kDa PrPres. More amounts of Poly A were also effective but SSpDNA was not, at both more or lesser amounts to precipitates of the 23-kDa molecule. Poly C and scBrh mixing to precipitate the 23 kDa PrPres was not proved effective as Poly A or SSpDNA in the ASP. The three poly nucleotides were not bound to the primary and secondary antibodies (Fig. 5 Panel II lanes 11–16).
Discussion
ASP is an easy and rapid method to examine the presence of PrPres in the blood. The method appeared to be more effective when blood samples rather than brain homogenates were used. The reason for this finding is not proven. One possibility is that PrPres may self-aggregate, and thus, precipitate by this method. Another possibility is that some other substances may play a role in the precipitation reaction. We previously reported that mixing of scBrh with mcPl resulted in the appearance of novel antiPrP mAb-reactive protein bands, in a manner similar to the scPl-derived bands yielded by ASP. 26 This observation suggested that the PrPres may be associated with additional partner substances in the blood of infected animals and hence can be precipitated. However, the substances that may become associated with the PrPres in the blood of infected animals have not yet been identified.
Several substances have been reported to have affinity to PrPres. Of these substances, Plsmg and hemin are known to be the possible carrier substances for PrPres in the blood.14,19,27 HepSulp has already been reported to be a possible PrPres receptor substance.17,18 Furthermore, botanic sulfated polysaccharides, such as pentosan polysulfate (PPS), have been reported to inhibit the binding of PrPres to the assumed receptor. 17 However, these substances were found to be somewhat unsatisfactory carrier substances when their affinity association with PrPres was tested using the ASP method.
Research for developing blood tests aimed at detecting PrPres molecules in its free or aggregated form by capturing specifically bound substances, such as mAbs or other molecules.13,28–30 However, irrespective of these efforts, none of the studies have yet been successful in meeting the final objective, presumably because it is extremely difficult to detect trace amounts of components, such as PrPres or its aggregates, when they exist within an enormous amount of contaminants. Furthermore, since even non-infected healthy individuals possess the PrPres-like PK-resistant PrP molecules or its aggregates, it is somewhat difficult to discriminate infected and healthy individuals on the basis of PrPres detection.31,32
On the other hand, according to the protein-only hypothesis, the PrPres aggregates are suspected to be the infectious entities. Therefore, it has been assumed that the presence of this protein or its aggregates is associated with infectivity or other related biological activities. One of the predictions of this hypothesis is that a 25-nm diameter particle is the most infective particle.33,34 It is not known whether this 25-nm particle is a homologous aggregate of the PrPres molecule or a heterogeneous aggregate of substances that are unrelated to PrPres. Furthermore, it is also unknown whether this particle was the suspected PrPres in the blood of infected individuals or not. On the other hand, observations which found a strong association between nucleic acids and the PrPres molecules were reported. These reports showed a correlation between the PrP molecule and viral replication having both positive and negative effects. The knocked out of the PrP gene (Prnp-/-) enhanced the poliovirus replication; whereas, transfection of the gene into the Prnp-knocked out cells suppressed the replication of coxsackievirus.35,36 The PrPc gene has been shown to have similar effects on the promoter function of retroviral coat proteins.37–39 These observations suggest that there is a direct interaction between the PrP molecule and viral nucleotides, although the effect of the molecule to viral nucleotides was not uniformly assumed. In addition, it has been reported that nucleotides (DNA and RNA) accelerated the process of PrP molecule assembly into amyloid fibrils, suggesting a direct correlation between PrPres and nucleotides. 40 Together with the report that the minimum requirement for the PMCA method included poly-anions (including RNA), the correlation of nucleic acids to the PrP molecule will prove to have important biological implications.41,42 It was suspected that the molecular site responsible for performing the biological activities of PrPc was its N terminus (especially, octa-repeat moiety); hence, it seemed uncertain that PrPres aggregated and yet retained its biological activities. 43 The relationship between the affinity association of nucleotides with PrPres and the biological function of the nucleotide-associated PrPres molecules are important issues that need to be addressed.
Since the association affinity of PK-mcPl with PrPres in scBrh was diminished by deglycosylation, we suspected that the polysaccharides of some glycoprotein component in the PK-mcPl played a role in the association, as has been reported previously. 26
As we experienced in our experiments, dilution of the scBrh with TBS-SDS resulted in almost diminished on the 30 kDa band and the smaller recovery of the PrPsc than expected even used the same volumes of original one. So, if we compared the observed patterns of PrPsc in standard lane of PK-scBrh on Figure 1 to that on Figures 2–5 electrophoresis patterns were quite different each other especially on their Mw and the intensity of the PrPsc signals on Figures 2–5 were unexpectedly low. It must be considered that some self aggregation of the PrPres occurred in non-diluted PK-scBrh but the aggregation might be dissociated in the diluted preparation using TBS-SDS. Therefore, the observation of mixing effects indicated on Figures 2–5 could be the results of joining effect with restitution and enhancement of complex formation between the PrPres and the partner substances.
Deglycosidation of the mcPl led to a decrease in its affinity to PrPres, although such an effect was not observed in the other glycoproteins examined. Deglycosylation of Plsm and Plsmg enhanced their association affinity, but no such change was observed in the case of Fet. Thus, we could not accurately confirm the role of saccharide chains on the glycoprotein between PrPres and aggregates. As deglycosidation buffer also has a strong denaturing property, the observed phenomenon was suspected rather than due to the denaturing than deglycosylation of Plsm and Plasmg. Although treatment of Poly A in deglycosylation buffer mixture resulted in an extreme enhancement of its affinity to PrPres, similar treatment did not affect the affinity of SSpDNA. However, under denaturing conditions, the affinity of both Poly A and SSpDNA was greatly enhanced. The affinity of Poly C was moderately affected under the both deglycosidation and denaturing conditions. Therefore, the affinities of the molecules were affected by both denaturizing and the presence/absence of PNGase F protein. The fact that denaturizing of the nucleotides resulted in an extreme enhancement in the association with PrPres suggested that the linearity of the nucleotides, especially for Poly A and SSpDNA, but not for Poly C, affected their interaction with PrPres. And the presence of protein in the denaturalizing buffer could not acquire the ability of enhancement for SSpDNA. This observation suggested that PrPres might affect some cellular functions by direct interaction with linear Poly A RNA. This presumption also leads to another assumption, if the ASP method is related to capture the biologically meaningful complex which appeared as the PrPres effect on cell function.39,41,42,44 If the affinity association of PrPres and Poly A RNA in the linear form has some role(s) in the infectivity of PrPres in TSE cases, then this might be a major issue. Using the acidic SDS precipitation method, we were able to infer that Poly A RNA acts as a carrier molecule and is effective in the detection of PrPres. It is also suspected that Poly A may be efficiently used to form a mimic of a PrPres aggresome in vitro, as has been suggested by Goggin, et al. 21 Our method also allows the identification of some other substances that might have an affinity to PrPres in vitro.
Conclusion
Denatured polynucleotide, especially Poly A, has the ability to bind to the PrPres molecule with high affinity to form aggregates. Formation of this aggregate enabled us to precipitate the PrPres molecule effectively by the acidic SDS precipitation method. These observations together with the assumption that the PrPres molecule is similar to the Poly A-binding protein, the Poly A-mediated precipitation of the PrPres through the acidic SDS precipitation method may be a reasonable method for the discrimination of TSE-infected animals from uninfected ones.
Abbreviations
Transmissible Spongiform Encephalopathy;
variant form Creutzfeldt—Jakob disease;
Bovine Spongiform Encephalopathy;
normal isoform of prion protein;
disease-related Proteinase K-resistant prion protein;
test method to diagnose the TSE using blood or its component;
Acidic SDS Precipitation;
monoclonal antibody;
horse-radish peroxidase-conjugated goat anti-mouse IgG;
Proteinase K;
Peptide-N-Glycosidase F;
Plasmin;
Plasminogen;
Heparan Sulfate;
Fetuin;
20 mer of synthetic poly A RNA;
20 mer of synthetic poly C;
Sheared Salmon Sperm DNA;
scrapie sc237-infected hamster plasma;
mock infected hamster plasma;
scrapie sc237-infected hamster brain homogenate;
PK-pretreated (50 μg/ml);
25 mM Tris-buffered saline (pH7.2);
TBS containing 3% SDS and 2 mM EDTA;
TBS containing 0.05% of Tween 20;
buffer mixture to deglycosidate the glycoprotein;
buffer mixture for denaturing the nucleotide;
mixture containing 0.15 M NaCl, 0.01 M acetic acid and 1 mM EDTA;
Poly vinyliden fluoride;
Protein misfolding cyclic amplification.
Publish with Libertas Academica and every scientist working in your field can read your article
“I would like to say that this is the most author-friendly editing process I have experienced in over 150 publications. Thank you most sincerely.”
“The communication between your staff and me has been terrific. Whenever progress is made with the manuscript, I receive notice. Quite honestly, I've never had such complete communication with a journal.”
“LA is different, and hopefully represents a kind of scientific publication machinery that removes the hurdles from free flow of scientific thought.”
Available to your entire community free of charge
Fairly and quickly peer reviewed
Yours! You retain copyright
Footnotes
Acknowledgment
We thank Drs. Y. Yokoyama and M. Takata, Research Center of Prion Diseases, National Institute of Animal Health of Japan, for their kind gift of the scrapie (sc237)-infected hamsters. We also thank Dr. T. Onodera, Molecular Immunology, Agriculture and Life Sciences, Tokyo University, for his useful discussion and support. We thank Drs. H. Okazaki, Director, Department of Infectious Disease Research, Central Blood Institute, the Japanese Red Cross Society, and Dr. M. Nishimura, one of our colleagues, for their valuable discussions. We are grateful to all the members of the Japanese Red Cross Society for their kind support and discussions.
The authors report no conflicts of interest.