
Abstract
Microbes are the most abundant biological entities found in the biosphere. Identification and measurement of microorganisms (including viruses, bacteria, archaea, fungi, and protists) in the biosphere cannot be readily achieved due to limitations in culturing methods. A non-culture based approach, called “metagenomics”, was developed that enabled researchers to comprehensively analyse microbial communities in different ecosystems. In this study, we highlight recent advances in the field of metagenomics for analyzing microbial communities in different ecosystems ranging from oceans to the human microbiome. Developments in several bioinformatics approaches are also discussed in context of microbial metagenomics that include taxonomic systems, sequence databases, and sequence-alignment tools. In summary, we provide a snapshot for the recent advances in metagenomics approach for analyzing changes in the microbial communities in different ecosystems.
Introduction
In the natural environment, constant polymicrobial interaction(s) occur between bacteria, viruses, protozoa, protists, archaea and fungi. These microbes do not exist in isolation and are often found in a dynamic “consortia” of different microbial species populations. 1 Understanding microbial population dynamics in a consortium will benefit the genomic information of all coexisting members. Isolating and sequencing the genome of an individual organism from a consortium might not be adequate as the single isolate cannot be a representative of the full genetic and metabolic potential of its associated members. Moreover, achieving culture conditions for isolating a single member from a consortium would be a daunting task. Traditional microbiologists were always dependent on culture-based techniques for the identification of microbes in environmental samples. The challenge of identifying uncultured organisms was totally ignored. However, an explosion of knowledge in the field of microbial physiology and genetics happened during 1960s to mid-1980s wherein some scientists came to believe that cultured microorganisms did not represent the whole microbial world. This was evidenced by the “great plate count anomaly” showing discrepancy in the microbial numbers between dilution plating and microscopy. 2 From then on, several independent studies supported the rise of this uncultured world of microbes. 3
New non-culture based approaches have recently been developed that can be extensively used for comprehensive analysis of different communities in a microbial consortia.1,4–6 Metagenomics or genomic studies of microorganisms refer to an non-culture based approach for collectively studying sets of genomes from a mixed population of microbes. 1 The term “Metagenomics” was first coined by Handelsman and his colleagues in their study of natural products from soil microbes. 1 Community genomics, environmental genomics, and population genomics are often used as synonyms for metagenomics. The field of metagenomics was initially started by an idea from Pace in 1985 7 that subsequently lead to several studies, starting from the first cloning of DNA directly from environmental samples in a phage vector, 8 and culminating in the direct random shotgun sequencing of environmental DNA.9,10 Since the use of the metagenomics approach in these independent studies, several other studies have come to use this approach to study microbial populations in a wide range of samples ranging from the oceans to humans.8,11–22 Metagenomics has also provided significant information on the “changes” in the microbial community. For example, using a metagenomic approach, studies have elucidated changes in the microbial composition in humans fed on different diets.22,23 Similarly, metagenomics have provided information in the changes of microbial composition in ticks collected from different geographic regions. 15
Metagenomic studies can be grouped into four categories based on different screening methods: (a) shotgun analysis using mass genome sequencing; (b) genomic activity-driven studies designed to search for specific microbial functions; (c) genomic sequence studies using phylogenetic or functional gene expression analysis; and (d) next generation sequencing technologies for determining whole gene content in environmental samples.4,6,18,24–28 These four methods can be sub-classified under unselective (shotgun analysis and next generation sequencing) and targeted (activity-driven and sequence-driven studies) metagenomics.4,6,15,24,26,28,29 Some studies have used an unselective metagenomic approach extensively because of its cost-effectiveness and simplicity in DNA sequencing. 30
In this review we summarize recent advances in the field of metagenomics in studying changes in bacterial and viral communities from different ecosystems, provide a snapshot of metagenomic analysis and applications of the metagenomic approach, and discuss the development of some of the approaches to answer the challenges faced in accessing metagenomic data.
Experimental Design for Metagenomic Analysis
A common sequence-based metagenomic approach involves steps that are outlined in Figure 1. Due to high experimental costs incurred in metagenomics projects, there is a definite requirement of proper experimental design with appropriate replication and statistical analyses. For example, the approximate cost to produce metagenomic data from one gram of soil requiring 6000 HiSeq2000 runs would cost $ 267 million. 31 A proper experimental design should ideally start with a question rather than technical or operational restriction. As the ultimate aim of metagenomic projects is to link functional and phylogenetic information of microbial communities to the chemical, physical, and other biological parameters that characterize the environment, suitable reference samples for comparison should be considered and emphasized in the experimental design. The biological or technical variations that may arise during the experiment should not be neglected and should instead be considered carefully in planning the experiment. As microbial systems are dynamic, temporal sampling from an environment can have substantial impact on data and interpretation. Proper replicates need to be included in the experimental design and should also consider the level at which replication takes place. In summary, a well-planned experimental design in metagenomic projects would facilitate integration of data sets into new or existing ecological models. 32
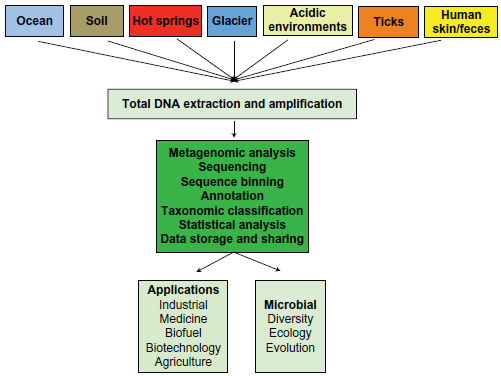
Overview of metagenomic analysis.
Sample Processing for Metagenomic Analysis
Sample processing is the first step of any metagenomic project. The DNA that will be used for metagenomic analysis should be representative of all cells present in the sample and should be ideal for generation of genomic libraries. High quality DNA extractions that include robust DNA extraction procedures are now readily available.10,32,33 Some of the common DNA extraction procedures, such as use of fractionation or selective lysis for isolating target DNA associated with a host,10,32–34 physical separation, and isolation of cells from the samples (eg, soil samples) or Direct lysis of cells in the soil matrix, have been reported. 33 Metagenomic analysis requires high nanogram to microgram amounts of DNA.33,35 In the case of samples that yield less DNA, amplification methods for the DNA is recommended. Multiple displacement amplification using random hexamers and phage phi29 polymerase has been reported to successfully amplify femtograms of DNA in order to produce micrograms of product.36,37
Metagenomic Sequencing
The metagenomics approach was originally focused on bacterial communities, but since been used to explore a wide range of microorganisms.1,5,20 Recently, several methods including Shotgun sequencing have been extensively used in metagenomic studies. 38 In Shotgun metagenomics, DNA isolated from an environmental sample is randomly sheared, sequenced in short fragments, and reconstructed into consensus sequences. With this method, detection of several microbes that would otherwise go unnoticed in culturing techniques was successful in environmental samples. 38 With the recent development of next-generation sequencing (NGS) (both 454/Roche and Illumina/Solexa systems), the whole of metagenomic sequencing has shifted from Sanger sequencing technology.39,40 However, Sanger sequencing is still considered for sequences with large insert sizes and a read length exceeding 700 base pairs. 41 Emulsion polymerase chain reaction is performed to clonally amplify random DNA fragments that are then attached to microscopic beads when NGS is performed using 454/Roche sequencer. The Beads attached to DNA fragments are deposited into picotitre plate followed by individual and parallel pyrosequencing. In the case of the Illumina/Solexa system, DNA fragments are immobilized on a surface and then solid-surface PCR amplification is performed. The amplified DNA fragments are then sequenced using reversible terminators in a sequencing-by-synthesis process. 42
A typical bacterial metagenomic analysis of environmental bacteria survey requires the use of the whole 16S ribosomal RNA (rRNA) gene.1,4 However, due to the read length restriction in NGS procedures, most surveys are aimed at characterizing selected hyper-variable regions of the 16S rRNA gene.15,43,44 The primary and secondary structures of the 16S rRNA gene show nine hyper-variable regions flanked by relatively conserved regions.15,43,44 This property makes hyper variable regions of 16S rRNA gene an optimal species molecular marker. 45 Recent studies have shown comparable results between sequencing of hyper-variable regions and sequencing of a full-length 16S rRNA gene.46,47 Based on these studies, it is recommended to design oligonucleotides for the V1-V3 region or V4-V7 region for Archaea and the V1-V3 region or V1-V4 region for bacteria.
Metagenomic Sequence Assembly, Binning, and Annotation
The sequenced DNA fragments are then processed for assembly using one of the two strategies, either reference-based assembly (co-assembly) or de novo assembly. Software packages such as Newbler (Roche), AMOS (http://sourceforge.net/projects/amos/), or MIRA 48 can be employed to perform reference-based assembly. For de novo assembly, tools based on the de Bruijn graphs are created to handle very large amounts of data.49,50 In addition, two new assembly programs (Meta Velvet and Meta-IDBA) 51 have been developed to deal with the non-clonality of natural populations. The sequenced information is then processed for binning to sort DNA sequences into taxonomic groups that might represent individual or closely related genomes. Several algorithms employing different methods of grouping sequences have been developed, including but not limited to Phylopythia, S-GSOM, PCAHIER, TACAO, IMG/M, MG-RAST, MOTHUR, MEGAN, TANGO, CARMA, SOrt-ITEMS, MetaPhyler, PhymmBL and MetaCluster.52–63 These algorithms have been developed depending on the type of input data generated from metagenomic sequencing.
Generally, metagenomic sequences are annotated in two steps: (a) Feature prediction is performed by identifying characteristics of interest within genes; and (b) functional annotation is performed by assigning putative gene functions and taxonomic neighbors. Several tools such as MG-RAST, IMG/M, FragGen-eScan, MetaGeneMark, Metagene, and Orphelia have been developed for classifying sequence stretches as either coding or non-coding.52–58,64–70 BLAST-based searches are also used for potentially identifying any missing information from these programs. Some of the other tools that are employed for predicting non-protein coding genes are tRNAs, Signal peptides. and CRISPRs.71–73 Other primary online sources for obtaining annotated nucleotides sequence information include the International Nucleotide Sequence Database Collaboration (INSDC), the DNA Data Bank of Japan, the European Nucleotide Archive, GenBank, and the Sequence Read Archive (SRA). By mid-September 2010, the SRA had accumulated more than 500 billion reads consisting of 60 trillion base pairs available for download. 74 SRA contained 80% of the sequencing data from the Illumina GA platform, as well as 15% and 5% from the SOLiD TM and Roche/454 platforms, resepectively. 74 Functional annotation of the metagenomic data is a major challenge, as only a small percentage of metagenomic sequences are annotated.54,65,75 The sequences that cannot be annotated, either because they might simply reflect erroneous coding sequences, because they might be real genes but encode for unknown biochemical functions, or because they may not have homology to known genes, are all grouped as ORFans. 76 Additional reference databases such as KEGG, egg-NOG, COG/KOG, PFAM, and TIGRFAM are all available online tools that can be used to study functional properties of ORFans. 76
Statistical Analysis and Data Sources
A typical metagenomic project contains an enormous amount of data that needs careful evaluation using proper statistical methods. Primer-E-Package is a popular tool that can perform a range of multivariate statistical analysis. 77 This package includes generation of multidimensional scaling plots, analysis of similarities (ANOSIM), identification of the species, and identification of gene functions (SIMPER). There is also a web-based tool called Metastats that has been used in recent studies. 78 The Shotgun-FunctionalizeR package also provides several statistical programs to evaluate functional differences between samples. 29 Due to the increasing number of metagenomic studies, it is important to deposit large sets of metagenomic data into databases. Deposition of metagenomic data in centralized services would not only facilitate comparative analysis of different metagenomic data but also facilitate a new level of organization and collaboration among researchers. Services like IMG/M, CAMERA, and MG-RAST are three prominent metagenomic databases that are available for large-scale metagenomic analysis.54,57,75
Metagenomics to Study Microbial Diversity in Environment
In the last decade, several studies have used the metagenomic approach and provided comprehensive data on microbial communities in different ecosystems. It is estimated that, depending on the sample and methods used, the number of bacteria in soil may vary from 467 species to 500,000 species.19,79–81 Curtis and colleagues have speculated that bacterial content may range up to 4 × 10
6
/ton of soil and the numbers of bacteria are unlikely to exceed 2 × 10
6
in the sea.
19
These comparisons clearly suggest that microbial content is several orders of magnitude less in the sea in comparison to soil environments. The members of the archaeal phylum
Soil is one of the most challenging environmental sources to analyze microbial diversity. Several parameters of soil, such as particle size, permeability, porosity, water content, mineral composition, and plant cover, can influence microbial composition.35,85,86 In addition, other factors such as collection and storage of soil sample, DNA extraction methods, host-vector systems used for DNA cloning, and representative soil sampling, can also influence the results of microbial content.35,85,86 With the advent of various technical developments, several landmark studies have been performed using the metagenomics approach.10,12,20,29,83,87–89 By direct cloning into plasmid, cosmid, or BAC vectors, novel genes from soil microbes that encode enzymes and antibiotics have been discovered. 90 These genes share little homology with known genes, thus illustrating the enormous potential of soil metagenomics in isolating novel classes of genes. Some of the genes that were isolated from soil microorganisms include lipases, proteases, oxidoreductases, amylases, antibiotics, antibiotic resistance enzymes, and membrane proteins.21,87,91–93
Using a metagenomics approach several studies have provided a wealth of information on microbial diversity in extreme environmental conditions. Studies from Barns and colleagues have provided information on microbial diversity in hot spring environments.94,95 Archaea similar to
Although these studies indicate the important role of microorganisms in biogeochemical cycles, many details remain unclear; until we fully understand the nature of microbial diversity in different environments, this will remain as an important area of investigation.
Viral Metagenomics
The development of metagenomic approaches has revolutionized evaluation of viral particles in environmental samples. The results from more than 24 independent studies have already been published. 98 These studies highlight that 50% of viral sequences are “unknown”. Of the remaining 50% “known” sequences, many had low amino acid similarities to known viral proteins and thus represent an uncategorized group. 20 These findings suggest a more complex diversity of viral genomes in comparison to bacteria in environmental samples. This is consistent with the findings that 30% of the open reading frames in sequenced viral genomes are ORFans, compared to 9% ORFans from bacteria. 99 Despite these challenges, viral metagenomics have developed methods to catalogue viruses in environmental samples based on identifiable sequences. Full genome sequences of novel viruses that were identified from different environments have already been reported and assembled. 100 Based on genomic structure and taxonomic metagenomic analysis, some of the studies have linked viruses with their potential hosts.101,102
Over the past decade, several studies using metagenomics have provided a substantial amount of information in the identification of new viruses from human samples.103–106 Most of the infectious diseases caused by viruses were documented before the identification of their causative agent. For example, Egyptian literature from approximately 3700 BC provided information on poliomyelitis. However, the causative agent for this disease was identified as poliomyelitis virus in 1909 AD. 107 Similar descriptions of clinical conditions likely caused by Smallpox were found in ancient literature from India 1500 BC long before the isolation of the Variola virus.108,109 With the steady rise in the development of viral metagenomics, several novel viruses have been isolated within a short amount of time that are associated with disease outcomes in humans.98,104–106 Novel viruses including Borna virus, Arena virus, Paralysis virus, LUJO virus, Astrovirus as etiology of mink shaking syndrome, Simian hemorrhagic fever virus, and Klassevirus have been identified by metagenomic approaches as causes of diseases in humans and other mammals.110–116 In addition, a recent study has provided important information in the identification of several viruses in a public-health setting. 104 These studies highlight future perspectives on the use of metagenomic approaches for generating enormous amounts of data in the identification of unknown and potentially infectious agents to humans, all in a short amount of time. Recent metagenomic analysis also addressed changes in the viral communities in Cystic Fibrosis and compared them to those of non-cystic fibrosis individuals. 105 In addition, interest in tapping the vast novelty of viral genetic information, especially phages, has brought great attention to the use of metagenomics in this field. 101 Overall, metagenomics has provided substantial insights to virus–host interactions and viral diversity in different environments.
Tick Metagenomics
Ticks are medically important arthropod vectors that transmit pathogens causing various human diseases.
117
The advancement of metagenomic approaches has facilitated research in studying microbial communities associated with medically important arthropod vectors.15,118 Using 454/Roche and Illumina-based metagenomic sequencing, Carpi et al have evaluated pathogen load and microbiome in
Industrial Metagenomics
With the advent of the metagenomic approach to discover novel genes that encode various enzymes, antibiotics, photoproteins, and membrane proteins from environmental uncultured bacteria, several industries have shown interest in exploiting these resources for the development of commercially available compounds. Metagenomics has provided access to novel enzymes and biocatalysts that were not initially achievable by conventional cultivable bacteria.21,87,119–121 In fact, global sales for enzymes were estimated to be $ 2.3 million in 2003, a figure that includes sales of enzymes in detergents, food applications, agriculture/feed, textile processing, pulp/paper, leather, and production of fine and bulk chemicals. 122 In light of increasing energy costs, environmental pollution, public health hazards, and recent global economic crises, the discovery of novel enzymes from metagenomic approaches can be viewed both as an opportunity and as a necessity. For example, Diversa, the largest biotech company focusing on the commercialization of metagenome technologies, has constructed and screened for various nitrilase gene sequences isolated from diverse environmental libraries. 123 This nitrilase enzyme library was marketed to several fine-chemical and pharmaceutical industries. 123 In summary, metagenomics has played a significant role in the identification of several bioactive molecules that have attracted interest from both academia and industrial companies.1,87,119–121
Metagenomic Application to Study Human Gut and Skin Microbiome
Over the past decade, metagenomics have provided great insights to the human microbiome. Waddington used a metaphor and regarded microbiota as an essential “organ” of the human body capable of performing metabolic functions that human cells might not be able to perform.124,125 Several factors such as specific microbial species colonizing the gut, niches they occupy, time, space, factors unique to the environment of each human being such as different dietary needs, and interactions with host cells can all influence taxonomic composition of the human microbiome. Metagenomics have uncovered nearly 1000 human-associated microorganisms’ draft genome sequences, along with 3.3 million unique microbial genes derived from the intestinal tract of over 100 European adults.17,126,127 Analysis of intestinal microbial content of humans across various continents revealed that microbes were clustered in 3 groups that are termed as enterotypes.17,22,126,127 Metagenomics of the human gut microbiome also revealed interesting functions carried by microorganisms within the gut, ranging from its role in newly discovered signaling mechanisms, vitamin production synthesis, glycan production, amino-acid, and xenobiotic metabolism. Several studies have also reported that microbial composition of the human gut is greatly affected by genetic background, age, diet, and health status of the host.17,22,126–128 Differences in microbial content were seen in all age groups of human beings. Babies (breast fed and formula fed), healthy and malnourished infants, youngsters, the elderly, humans that were either lean or obese, and humans with inflammatory bowel diseases (IBD) showed differences in microbial composition.23,88,129–131 A metagenomic study from De Filippo et al showed that European children who consumed a diet high in animal protein, sugar, starch, and fat, and low in fiber showed differences in gut microbial content in comparison to children fed on vegetarian diet consisting of carbohydrates, fiber, and non-animal protein.
127
Interestingly, the microbiome of European children was enriched with
Recent studies have used metagenomic approaches in looking at the microbial diversity of the human skin.135–138 Skin serves as a good host of microbes that include both commensal and pathogenic bacteria. Determination of microbial diversity in skin revealed several interesting findings.135–138 Bacteria belonging to
Future Directions
Over the past decade metagenomics has undoubtedly benefited the scientific world in rapidly analyzing changes in microbial communities in different environments. Despite exhaustive research efforts, both in financial and intellectual terms, the underlying mechanisms of the relationship between the microbial communities to the environment or to human gut metabolism, aging, and disease remains unclear. Therefore, improvements in metagenomic techniques that involve functional microbiomic approaches need to be developed. In addition, development of novel metagenomic approaches that consider several geochemical parameters is highly warranted to evaluate the complexity of microbial population in extreme environments. Metagenomics has provided identification of several new microbial genes from different environmental samples.11,12,14,15,17,19,91,93,135 Heterologous gene expression is an important and challenging approach that is required to identify the function of new genes identified by metagenomic studies.139–142 Studies have successfully used a heterologous gene expression system to identify several antibiotic resistance genes.140,142–145
Author Contributions
Conceived and designed the experiments: GN, HS. Analyzed the data: GN, HS. Wrote the first draft of the manuscript: GN, HS. Contributed to the writing of the manuscript: GN, HS. Agree with manuscript results and conclusions: GN, HS. Jointly developed the structure and arguments for the paper: GN, HS. Made critical revisions and approved final version: HS. All authors reviewed and approved of the final manuscript.
Funding
This work was supported by independent start-up funds from Old Dominion University to GN and HS.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests. Provenance: the authors were invited to submit this paper.