
Abstract
Quality control and read preprocessing are critical steps in the analysis of data sets generated from high-throughput genomic screens. In the most extreme cases, improper preprocessing can negatively affect downstream analyses and may lead to incorrect biological conclusions. Here, we present PathoQC, a streamlined toolkit that seamlessly combines the benefits of several popular quality control software approaches for preprocessing next-generation sequencing data. PathoQC provides a variety of quality control options appropriate for most high-throughput sequencing applications. PathoQC is primarily developed as a module in the PathoScope software suite for metagenomic analysis. However, PathoQC is also available as an open-source Python module that can run as a stand-alone application or can be easily integrated into any bioinformatics workflow. PathoQC achieves high performance by supporting parallel computation and is an effective tool that removes technical sequencing artifacts and facilitates robust downstream analysis. The PathoQC software package is available at http://sourceforge.net/projects/PathoScope/.
Introduction
Advanced and efficient next-generation sequencing (NGS) technologies are currently providing unprecedented resources and insight for numerous applications in biology and biomedicine. These technologies are now regularly leveraged in unique and novel ways to characterize biological pathways, 1 understand disease etiology,2,3 discover novel drug targets,1,4,5 and develop personalized treatment regimes.6–8 However, data from these experiments present many difficult technical and computational challenges. For example, due to the extremely large size of data sets generated from these experiments, the handling, storage, and analysis of multiple massive data files is now routine work in the laboratory. 9 Furthermore, data from these experiments usually contain complex and high-dimensional artifacts that must be removed before the data can be properly analyzed. Identifying and removing low-quality sequencing reads is critical to the stability and accuracy of downstream analyses. For example, due to current limitations of NGS technologies, the fidelity of the sequencing “base calls” and confidence in sequencing read counts can be affected by several technical factors during the sample preparation, library preparation, and sequencing/imaging step.10–14 Failure to filter sequence duplications can negatively skew read abundance or expression measures or lead to false variant calling.15–18 Similarly, discrepancies among overlapped reads containing erroneous residues can complicate the assembly process or might lead to incomplete contiguous sequence extensions in genome assembly. 19
Proper and complete quality control (QC) procedures for preprocessing NGS reads typically comprise two essential components: (1) statistical evaluation of overall read quality or sequence composition, and (2) read cleaning, processing, and filtering. In general, the former helps users to determine the characteristics of data such as the distribution of the GC base content in the reads, the range and distribution of base-quality scores, and the overall levels of sequence ambiguity or complexity (repetitiveness). These statistical measures help evaluate the overall quality of the sequencing data set and aids in the selection of parameters and cutoff values needed in the filtering step. The read cleaning, processing, and filtering step includes removing “tag” sequences (adapters, primers, and barcodes) or low-quality parts of the read, apart from filtering entire sequences of low quality or sequences that are too short after trimming. This step may also require the removal of multiple duplicated reads or reads from unexpected genomic contamination from the experiment or library preparation.
There are many QC software packages available to filter or trim low-quality reads.20–23 However, these methods typically focus on only one or two of the QC steps and do not provide a complete QC workflow. Furthermore, data preprocessing is often separated from the main analysis workflow and may require the application of multiple QC tools and steps for each sequencing read sample in the data set. Overall, read QC is often a complex, daunting task that often slows down the entire analysis workflow.
Here, we present PathoQC, a comprehensive and user-friendly command line QC software for experienced computational scientists, which is designed to perform complete, high-quality preprocessing of sequencing reads in a single step. Our primary goal in developing PathoQC is to provide a flexible and simple user-friendly software module for QC preprocessing for most of DNA or RNA sequencing assays. PathoQC was originally released as a “plug-in” module for the PathoScope 2.0 metagenomics framework, 24 but it also functions as a stand-alone pipeline that can be easily integrated within other NGS analysis pipelines. At the heart of PathoQC lies the parallel processing module with paired-end (PE) reads support that integrates with three state-of-the-art QC modules, namely, FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc), Cutadapt, 20 and Prinseq. 21 PathoQC is designed to utilize the strengths of these approaches as well as provide integrative benefits not available when using each of the QC modules alone. In addition, the efficient parallel processing module of PathoQC decreases the amount of time necessary for QC and better utilizes the resources for cluster submission of workflows in which downstream analysis steps also require multiple central processing units (CPUs). Furthermore, PathoQC contains a couple of unique features such as handling valid singleton reads in PE inputs and a trimming option for either a higher-quality single-nucleotide polymorphism (SNP) analysis or a better alignment rate. Next, we describe the PathoQC workflow and compare it to other QC approaches in terms of processed data quality and computational time required for preprocessing. We illustrate the benefits by applying PathoQC to a metagenomics sequencing data set used for detecting pathogens that have previously been established to play a role in carcinogenesis. We explore effects of the QC processing steps on human RNA-Seq reads, and our benchmarks demonstrate that PathoQC leads to more confident and sensitive variant calling than other QC methods.
Methods
Patho QC work flow details
PathoQC integrates the three core software programs for QC analysis, namely, FASTQC, Cutadapt, and Prinseq. PathoQC utilizes the individual strengths of these programs to provide high-quality read preprocessing. For example, the Prinseq stand-alone version provides QC options related to base quality, sequence complexity, GC content, and sequence artifacts, but it does not detect and trim tag sequences. In contrast, Cutadapt has been successfully used to trim multiple tag sequences in numerous sequence libraries25–28 but does not have other features available in Prinseq. Furthermore, we use FASTQC to choose appropriate processing parameters to minimize user input if desired. By combining the strengths of these tools together and introducing novel features such as parallel computation and better handling of PE reads, PathoQC provides the most sophisticated QC workflow available (Fig. 1 for workflow). Note that PathoQC only takes care of the preprocessing step before alignments.

PathoQC module workflow. PathoQC is a read quality control software module that performs several read quality control steps, including detecting and trimming adapters, trimming low-quality bases at both ends of reads, and filtering low-complexity and duplicate sequences. PathoQC is an automated, parallel workflow that seamlessly combines the strengths of the Cutadapt, Prinseq, and FASTQC read preprocessing tools before any secondary analysis (eg, alignment or SNP analysis).
PathoQC consists of four steps. In Step 1, the user provides the sequencing read data set in FASTQ 29 or FASTA (http://www.ncbi.nlm.nih.gov/blast/fasta.shtml) format. Unless the user specifies input parameters in a run option, FASTQC would extract Phred offset, read length, a minimum base quality to trim, and primers/adapters among overrepresented sequences. In Step 2, PathoQC applies the FASTQC algorithm, produces the standard FASTQC visual output and results, and PathoQC also automatically collects the Phred offset, the minimum base quality, the range of read lengths, and overrepresented adapters or primers (if not provided in Step 1) for use in further preprocessing steps. In Step 3, PathoQC applies Cutadapt to remove overrepresented sequencing tags from the data. For all tag sequences provided by the user or from Step 2, Cutadapt performs an “end-space free alignment” in order of O(nk), where n is the total number of the characters in all reads and k is the sum of the length of the adapters. 30 Cutadapt can also simultaneously search for multiple adapters in a single run of the program. 20 Finally, it conducts a gapped alignment by considering homopolymertype artificial insertions and deletions (eg, pyrosequencing). In Step 4, Prinseq is used to trim low-quality bases and remove reads that are too short, of low complexity, or redundant. Depending on the platform generating the input reads (eg, Illumina), Prinseq trims lower-quality bases at the 5’ or 3’ ends of the reads 31 or removes reads largely contaminated with homopolymer-length sequencing errors such as “AAAA” or “TTTTTTTT” 32 (eg, pyrosequencing). Furthermore, the Prinseq software provides a large number of command line options for trimming sequence tags and filtering reads by their lengths, quality scores, GC contents, proportions of ambiguous base calls, sequence duplicates, and sequence complexities. 21 These options can be specified from the PathoQC command line arguments. Table 1 summarizes the options supported by PathoQC and compares these with other existing QC approaches. The following subsections detail other unique options and functionalities available in the PathoQC software.
Comparison of features for NGS quality control methods.

Parallel computation
PathoQC supports parallel computation with multiple threads across a compute node with multiple cores. To accomplish this, PathoQC uses two standard Python modules, multiprocessing and Queue. 33 In its parallel implementation, PathoQC calculates the read file size and evenly distributes the reads to multiple CPUs or threads (as specified by the user). The PathoQC pipeline is applied automatically to each subset and the processed reads are merged into one FASTQ file, for further processing. Neither Cutadapt nor Prinseq support parallel computation, but most downstream alignment and analysis steps utilize multiple threads. This means that the computational resources on the cluster, cloud, or local machine are left unutilized while the QC pipeline processes each sample individually on a single CPU. In contrast, PathoQC allows users to match CPU usage for the QC steps with downstream analysis needs, thereby providing more optimal usage of computational resources.
PE reads
PE sequencing is now a standard and very common sequencing approach. Most QC workflows completely separate a read pair from valid PE read set if one read is filtered by QC processing. In contrast, PathoQC will collect all high-quality “singleton” reads and merge them to a valid PE read FASTQ file format so that we can align them to a reference genome in a single run. This option can increase the overall mapping efficiency for PE reads. For instance, in gene fusion or structure variation study, keeping more discordant pairs may help in identifying chromosomal breakpoints.
Additional QC features
In addition to the QC features provided by the three core QC software packages, PathoQC provides users four additional features: 1) PathoQC provides a read summary report containing information, such as the range of the processed read lengths; 2) PathoQC supports Disk Operating System (DOS)-format FASTQ files; 3) PathoQC can automatically detect adapters or primers and, moreover, it determines a minimum base-quality cutoff adaptively for input reads; 4) in PCR deduplication, it retains the one with the highest base-quality reads among the identical products; and 5) PathoQC can retain low-quality bases (instead of trimming them) if the length of good-quality bases is longer than a minimum length that the user specifies. The final option can help increase the mapping specificity when an alignment program allow reads to be soft clipped by a Smith–Waterman local scoring scheme. 34
Results and Discussion
We compared the performance of PathoQC with that of four stand-alone QC software approaches, namely, Cutadapt, Prinseq, NGS QCToolkit v2.3 22 (hereafter QCToolkit), and QC-Chain. 23 These programs were chosen for comparison because they provide nearly complete QC preprocessing options similar to PathoQC. A comparison of the features provided by the five software packages is given in Table 1.
The experiments were conducted with a three-fold purpose. 1) Given three samples (two carcinoma cell line RNA samples and one metagenomic DNA sample), we evaluate each QC software's performance (speed, memory usage, the number of filtered bases, etc); 2) in the first case study, we evaluate the consequence of each QC preprocessing strategy on species identification with those preprocessed samples; and 3) in the second case study, six human RNA-Seq samples containing External RNA Control Consortium (ERCC) spike-in control 35 are used to explore the effects of QC software on gene expression and SNP analysis.
Data set descriptions
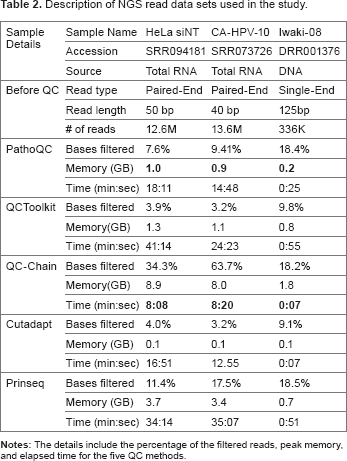
The first data set consists of 12.6 million, 50-base pair (bp) strand-specific, PE sequencing of RNA from HeLa cells 36 (SRR094181, HeLa_siNT). In this data set, we observed two Illumina PCR Primer Index sequences, one from each of the sequencing read pairs. The second sample contains 13.6 million, 40-bp PE sequencing reads from a human prostate cancer cell line 37 (SRR073726, CA-HPV-10). The third sample is from a metagenomics study containing 336K single-end DNA sequencing reads from a human abscess sample of unknown etiology 38 (DRR001376, Iwaki-08). A summary of data set features is given in Table 2. The fourth sample (study ID: GSE 49712 39 ) is described in the section on Case study II.
Description of NGS read data sets used in the study.
Evaluation of PathoQC in cancer metagenomics
QC results
We applied each of the QC programs (PathoQC, QCToolkit, QC-Chain, Cutadapt, and Prinseq) on all three data sets using Linux desktop with 16-gigabyte random-access memory by utilizing four CPUs. We set the base-quality cutoff as 3–5, which corresponds to min(Q phred )-2 for each data set (In the section Case study I, we also repeat the same experiment under a higher base-quality cutoff). We set the minimum read length satisfying the base-quality cutoff as 30 bp for PE reads and as 35 bp for single-end reads. There are no fixed cutoff values for those parameters. It rather depends on which analysis is of user interest. In the metagenomic samples, we consider a higher mapping rate so that it can help increasing sensitivity in identifying prevalent nonhost organisms. Duplicated reads are filtered if the pipeline provided this option. The QC parameters used for all programs and data sets are given in Table 3.
Parameters used in QC software and RNA-Seq analysis.

Computational performance
Overall, QC-Chain preprocessed data faster than the other two pipeline methods, completing the QC processing 1.8–3.6 times faster than PathoQC and 2.9–7.9 times faster than QC Tookit. PathoQC required less memory than the other programs, which needed 1.2–2.7 (QCToolkit) and 6.0–8.9 (QC-Chain) times more memory than PathoQC. Both Cutadapt and Prinseq were capable of utilizing only a single CPU, while PathoQC – with four CPUs – was nearly 2.5–3.5 times faster than the sum of the two elapsed times, indicating that the data-processing speed is linearly scalable with respect to the number of CPUs.
Filtering results
The three complete QC software packages filtered some uninformative reads after trimming, 3%–4% of the raw reads for HeLa siNT, 2%–3% for CA-HPV-10, and 9%–10% for Iwaki-08, respectively (Fig. 2A–C). QCToolkit does not filter duplicated reads. Only PathoQC and Prinseq remove identical sequence copies, including reverse complementary sequences. QC-Chain marked 30% and 60% of reads as duplicates in two PE read data sets, which is seven to eight times larger than the results with PathoQC.

The fraction of filtered reads in each step of QC. (
Cutadapt was only able to trim input reads, showing similar results overall with that of QCToolkit for all three samples. On the other hand, the number of filtered reads after the trimming step was not distinguishable between PathoQC and Prinseq, except for the first sample, wherein Prinseq did not remove primers, so more reads were retained. In addition, PathoQC filtered out reads of low sequence complexity. Such sequences can produce a larger number of high-scoring but biologically insignificant results in database searches. This feature is very useful for metagenomic data, which will be discussed shortly. Particularly, in Iwaki-08, it removed 4.1% of the reads having lower sequence complexity.
case study I
We also evaluated the downstream impact of the QC procedures on downstream applications. After preprocessing the reads, we used the PathoScope 2.0 software 40 to align and profile microbial content in these samples. Specifically, we examined the impact of read QC on the ability to identify cancer-causing pathogens. We analyzed the two samples CA-HPV-10 and Iwaki-08 after preprocessing them by each of the QC programs. We used PathoScope (refer Table 3 for parameters used) to align the two QC read samples against both the human reference genome and a reference genome library containing all viral and bacterial genome sequences. All reference sequences were obtained from the National Center for Biotechnology Information (NCBI) nucleotide collection (nt) database as of September 2013. The results are illustrated in Figure 2C.
PathoScope species identification
PathoScope removed potential human sequencing reads from these data, leaving 4,242 nonhost reads after PathoQC, 7,112 reads after QCTookit, 1,109 reads after QC-Chain, 7,107 reads after Cutadapt, and 3,557 reads after Prinseq. In the reads preprocessed by PathoQC, PathoScope ranked human papillomavirus 18 (HPV18) as the most prevalent microbe, assigning 79% of the nonhost reads (160 x coverage) to this pathogen. The Prinseq QC reads resulted in 75% assigned to HPV18 (127 x coverage), QCToolkit and Cutadapt assigned 59% of the reads to HPV18 (200 x coverage), and QC-Chain assigned HPV18 as the second most abundant pathogen, with only 16% of the reads (9.1 x coverage) assigned to HPV18. We also used SAMTools 34 to generate contiguous aligned sequences (contigs) for the reads assigned to HPV18. The N50 for all QC programs was 833 bp, except for QC-Chain, which had an N50 of 307 bp. Thermoanaerobacter wiegelii Rt8.B1 was also highly ranked by PathoScope with 28% of the reads from QCToolkit and Cutadapt. This species was not identified in the data processed by the other tools (the estimated proportion in the QC-Chain was negligible, 0.7%). Upon closer inspection and reference-guided assembly with SAMTools mentioned above, we observed that all contigs corresponding to this species consisting of consecutive As or Ts, suggesting that it is a false-positive result. In the Iwaki-8 data set, we observed improved PathoScope results for the PathoQC, Cutadapt, and Prinseq data, with 72%–73% of the reads sequenced from a pathogen infecting the sample being from Francisella tularensis subsp. holarctica FSC200. The other methods performed well overall but assigned fewer reads to this species.
For the PE RNA-Seq reads from HeLa cell line, QC-Chain removed all paired reads (30%) due to the presence of one read in the pair with low complexity. For the prostate cancer cell line, QC-Chain removed almost 60% of reads in its duplicate-filtering step, whereas PathoQC filtered only 7.7% of reads because it considers both reads in the pair simultaneously. This demonstrates one of the most important novel features of the PathoQC approach.
Higher phred quality cutoff
The cutoff can be chosen depending on the overall read quality, the read length, and the sequencing platform used. In the previous experiments, eg, we applied a less stringent base-quality cutoff so that it can keep as many bases in the reads as possible, which can increase the mapping specificity when the input read length is short. 41
The same experiments were repeated with a higher read quality cutoff (20 in Phred score for the sample CA-HPV-10). The overall evidence score for the pathogen by PathoQC becomes higher. PathoQC filters out 9.44% of the reads and the identification result remains nearly the same as the previous result, reassigning 77.1% read proportion to HPV18 in the first place. Two other QC programs, NGSToolkit and QC-Chain, filtered 6.67% and 82.8% of the reads, respectively. With these refined reads, PathoQC identifies HPV18 at the first and second ranks from top, with 60.3% and 17.5% read proportions, respectively.
Case study II
It is of great interest to assess the impact of QC data preprocessing on differentially expressed (DE) transcriptome analysis or SNP identification. Numerous quantification algorithms have been developed to statistically capture real mRNA levels within the cells for discriminating among different phenotypes.39,42 Recently, a study has shown that trimming tends to increase the quality and reliability of the analysis while reducing the computational resource requirement. 43 Depending on the read quality, some optimal parameters are suggested. 41 The QC software is evaluated with an RNA-sequencing data set containing the ERCC spike-in control.35,44 The objectives in the following discussion are to observe how QC influences 1) RNA-Seq alignment sensitivity, 2) relative DE abundance measurement with ERCC, and 3) SNP callings’ sensitivity and accuracy.

The distribution of nonhost genomes inferred by Pathoscope for the three QC pipelines in the Ca-HPV-10 data set. In this example, viral and bacterial genomes were included in a target reference genome library. Human papillomavirus 18 (HPV18) ranked highest in reads preprocessed by PathoQC (79%), Prinseq (75%), Cutadapt (59%), and QCtoolkit (59%). The pathogen ranked second for reads preprocessed by QC-Chain (18%). T. wiegelii, which is a false-positive genome found in the reads by QCtoolkit and QC-Chain, was not found in either the PathoQC- or Prinseq-processed reads.
Data description
From a previous study GSE49712, 39 six RNA-Seq samples (out of 10 technical replicates) were randomly selected. The six samples were grouped into two conditions with respect to ERCC spike-in control mix ratio. Each sample contained 60–111 million 101-bp PE RNA-Seqs from the Illumina platform (Fig. 4).

The number of properly paired alignments. The six 101-bp-long Illumina PE RNA sequencing reads randomly selected from a previous study (GSE49712) were preprocessed under equivalent quality-control parameters and a total of 24 reads, including the case of skipping any quality control (ie, “WithoutQC”) were aligned by TopHat2 in the same configuration. Refer to the main text for the parameters. The y-axis indicates the total number of properly paired or concordant alignment reads, except that “original” indicates the total number of raw reads. Across all replicas, reads preprocessed by PathoQC generated 5%–7% more confident alignments than the other methods.
For each RNA-Seq sample, PathoQC, NGSToolkit, and QC-Chain were run under the same parameters used in Table 3, except that 1) all duplicated reads were retained for all methods and 2) setting “-v min” in PathoQC parameters trims low-quality bases at the 3’ ends of the Illumina reads.
Effect on gene expression analysis
Twenty-four PE reads (including 18 QC reads and 6 raw reads) were mapped to the human genome (hg19/GRCh37) using TopHat (v2.0.9) 45 with the same parameters used in a previous study 39 (Table 3).
From our experiment results, QC preprocessing would not improve the alignment accuracy in terms of how closely the relative ratio of two samples’ read counts mapped to the ERCC sequences are correlated to the ERCC mixing ratio in control. We observed that the difference in the mapping counts on ERCC reference between two conditions is nonnegligible (1.01%–1.14% for samples SRR950078, SRR950082, and SRR950086 under the condition A vs 1.27%–1.92% for samples SRR950079, SRR950083, and SRR950087 under the condition B). Given the alignment files (BAM), HTSeq v0.6.0 46 was used to generate the count matrix as shown in Table 3. Then, the count matrix was normalized with the mean value of the hit counts across the gene sets. Table 4 represents the correlation coefficients between the predicted ERCC mix ratio and an actual control ratio. The results varied depending on the sample sets (eg, in the first two groups of samples, QC process improved the correlation but not in the last sample).
The correlation coefficient of the ERCC alignment ratio between two condition samples to the actual ERCC RNA spike-in control mixing ratio.

Higher confident mappings
PathoQC increases the alignment rate. Taking into account the mapping accuracy, it relies on the number of properly paired alignments (or equivalently concordant pair alignments), meaning that both ends of the read were mapped and they were mapped within a reasonable fragment length from the library preparation. In Figure 4, the PathoQC alignment rate ranges from 76% to 86% of the total number of original reads, which is 5%–7% higher than the other methods (including the case of bypassing any QC, henceforth termed “WithoutQC”).
SNP analysis
PathoQC improves the overall genotype quality. For each alignment file, we run sam-stats (https://code.google.com/p/ea-utils/) to compute both base quality and the percentage of reads containing at least one mismatch against a reference genome sequence (Fig. 5). As expected, both qualities improve after the implementation of any QC. In particular, PathoQC trims the consecutive lowest bases at the 3’ ends in the Illumina RNA-Seq so that it can achieve the best base quality compared to the other QC methods, which retain original reads but only filter out reads containing mostly low-quality bases.

The quality analysis on the aligned reads in GSE49712 using sam-stats. (
PathoQC helps in discovering more SNP calls than the other methods. An assembly- and haplotype-based SNP caller (Platypus 47 ) was run on 24 Binary Sequence Alignment/Map format (BAM) files with highly confident calling parameters (Table 3), meaning that it relies on all highly confident mappings and base calls. In SRR950078, for instance, a VCF (variant call format) file derived from PathoQC contains 43,694 SNPs.
In Figure 6A, all three methods except PathoQC show more commonality (for instance, 40,345 SNPs shared by NGSToolkit and WithoutQC vs 39,090 SNPs shared by PathoQC and NGSToolkit). This indicates that PathoQC does not call approximately 1,000 of the SNPs that are called by all the others. Nonetheless, PathoQC discovers 2.5 times more unique SNPs than the other methods.
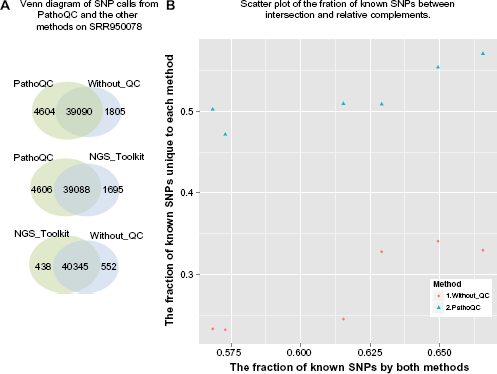
The effect of quality control on SNP analysis. An assembly- and haplotype-based SNP caller, Platypus, runs on 24 BAM files with a highly confident calling condition. Refer to the main text for the options. Note that we consider both heterozygous and heterozygous sequences. (
For all samples, the quality of the variant calls is examined by comparing them to the previously known SNP database. Among SNPs uniquely called by PathoQC, the fraction of known SNPs (ie, the variants reported into NCBI dbSNP human build 142) is 0.55, which is more comparable to the known SNP rate (0.58–0.65) commonly shared by two methods than it is to the fraction of known SNPs unique to WithoutQC, 0.3 (Fig. 6B). It suggests that the SNP detected by our QC helps downstream analysis achieve a better quality of variant calls than the other methods.
It is interesting to note that TopHat2 only supports end-to-end mapping and thus Illumina reads with potentially random matches or mismatches at 3’ ends are not allowed to map. For this reason, PathoQC is designed to balance sensitivity and specificity of alignment by trimming 3’ ends with minor base-quality cutoff and by retaining a high quality of singleton reads. Therefore, PathoQC helps to achieve the highest mapping sensitivity and it consequently facilitates the discovery of more private SNPs in the RNA-Seq data.
Conclusion
The objective of QCs in high-throughput sequencing reads is to monitor the quality of reads and to filter out sequencing artifacts, contamination, and unacceptable quality recurring in each NGS platform. Depending on which sequencing platform is used, how sequencing libraries are prepared, which features are available to the NGS aligner that a user employs, what is the main application (eg, species identification, differentially expressed transcriptome analysis, or a high-sensitive SNP analysis, etc), it is highly desirable to prepare input reads to achieve the best result. For this reason, QC software that can provide rich customized options is preferable.
PathoQC offers the most comprehensive QC features to improve the quality of downstream results from sequencing studies. PathoQC combines the strengths of three commonly used QC tools (FASTQC, Cutadapt, and Prinseq), along with several novel features, into a complete QC software module. The PathoQC pipeline consists of three major steps: 1) FASTQC is utilized to evaluate the overall quality of the data set and to identify QC parameters such as the Phred offset and overrepresented sequence tags; 2) Cutadapt is used to remove sequence tags; and 3) Prinseq is used to remove low-quality bases/reads and reads that have low complexity. PathoQC uses an efficient, parallel implementation that increases processing speed and better utilizes server resources. The software module is constructed for easy integration into any bioinformatics workflow. Under multiple experimental conditions, we showed that the PathoQC pipeline achieves excellent scalability and high-quality results.
In metagenomic samples, experimental results showed that PathoQC provides several important QC features, including filtering duplicated and low sequence complexity reads, which improved the quality of the predicted pathogen identification compared to other QC methods. In ERCC RNA spike-in control mixture samples, PathoQC's strategy of handling RNA-Seq (eg, trimming and retaining singleton reads) improves an alignment's quality in terms of both sensitivity and accuracy, as well as facilitating SNP identification.
Author Contributions
Conceived and designed the experiments: CJH. Analyzed the data: CJH, WEJ. Wrote the first draft of the manuscript: CJH, WEJ. Contributed to the writing of the manuscript: CJH, WEJ, SM. Agree with manuscript results and conclusions: CJH, WEJ, SM. Jointly developed the structure and arguments for the paper: CJH, WEJ. Made critical revisions and approved final version: CJH, WEJ. All authors reviewed and approved of the final manuscript.
Footnotes
Acknowledgments
This research was conducted using the Linux Clusters for Genetic Analysis (LinGA) computing resources at the Boston University Medical Campus (Boston, MA, USA).