
Abstract
Accurate diagnosis of chronic, non-healing wounds is challenging and time-consuming because it can be caused by a variety of etiologies. This brief report presents an unusual case of a chronic wound lasting for 10 months investigated by deep metagenomic sequencing. Epstein-Barr virus (EBV) was identified in the wound and subsequently validated by in situ hybridization. Histopathologic examination eventually revealed that the non-healing wound was due to an EBV-associated NK/T cell lymphoma. By identifying mutations across the viral genome, the virus was classified as Type I EBV and clustered with others of geographic proximity. Our results suggest that metagenomic shotgun sequencing can not only rapidly and accurately identify the presence of underlying pathogens but also provide strain-level resolution for the surveillance of viral epidemiology.
Background
Chronic, non-healing wounds can be caused by a variety of diseases, ranging from vascular disease, infection, diabetes, and metabolic disease 1 ; thus, accurate diagnosis is very challenging and time-consuming. In recent years, metagenomic whole-genome shotgun (mWGS) sequencing has emerged as a new technology for the diagnosis of pathogens without the need for culture.2-4 Several studies have used metagenomics for identifying pathogens in diabetic foot ulcers, 5 skin and soft tissue infections. 6 However, the concordance rate between mNGS and culture validation is still inadequate. 7
The major challenge of applying mWGS in clinical diagnosis is the enormous amount of contaminating DNA from the host. In order to reduce host DNA, specific PCR can effectively enrich the pathogens of interest, but this is only applicable when targets are known.8,9 The adaptive sequencing of Oxford Nanopore platforms can theoretically remove host DNA during sequencing. 10 However, the technology is still too expensive and the accuracy is insufficient for clinical usage. Herein, we combine deep metagenomic sequencing with analysis techniques for the identification of underlying pathogens within a chronic wound.
Methods
Patient presentation, sampling, and sequencing
A 55-year-old woman was admitted to the hospital because of painful ulcerating lesions over right lower third medial shin accompanied with fever for 9 months. The ulcerating wound evolved despite multiple debridement and antibiotics treatment. She underwent below-knee amputation 8 months after the onset of this condition. Due to poor wound healing, above-knee amputation was done in 10 months. The tissue was sampled during surgical debridement, which was done before amputation and fixed in 10% buffered formalin.
The tissue sample was firstly grounded and mixed with 1 g of 0.5-mm diameter glass beads and then placed on a vortex mixer for 30 minutes at 3000 rpm. DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA, USA) was used for DNA extraction in 300 μl of the sample following the manufacturer’s instructions. We used an enzymatic method to fragment the DNA into 150-200 bp in length. The DNA library was built through end-repaired adapter and polymerase chain reaction amplification. We applied the DNA Qubit Assay (Thermo Fisher) to determine the DNA concentrations and used an Agilent 2100 system (Agilent Technologies, Santa Clara, CA) to evaluate DNA quality electrophoretically. The DNA library was built through end-repaired adapter and polymerase chain reaction amplification using MGIEasy FS DNA Library Prep Kit (MGI). We then transformed the single-strand circularized DNA library into DNA nanoballs (DNBs) and sequenced by DNBSeq-G50 with average read length equal to 50 bp.
Bioinformatics analysis
The sequencing reads were preprocessed by removing low-quality (ie, reads <80% phred score Q30), duplicated, and reads shorter than 35 bp in length. The remaining high-quality reads were BWA-aligned against the human genome (hg38) to remove human-derived sequences. 11 The non-human reads were BWA-aligned to the NCBI microbial reference genomes (RefSeq) for taxonomic classification. The species of lower read counts are considered as reagent/environmental contamination or alignment errors due to short-read mapping ambiguity.
In order to classify the EBV into type 1 or type 2, the viral reads were 6-frame translated and mapped to 6 type-differentiating protein sequences by Diamond: EBNA-1, EBNA-2, EBNA-3A, EBNA-3B, EBNA-3C, and LMP-1. 12 The average nucleotide identity of our strain with respect to others was computed by mapping reads onto the corresponding EBV genomes and parsing the alignment (CIGAR) via custom scripts.
Ethical approval
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board in Taichung Veterans General Hospital (No. CE20004B).
Results
Metagenomic sequencing and validation of EBV in the chronic wound
We applied deep metagenomic sequencing (168 million reads) to the gDNA extracted from a chronic wound tissue lasting for 10 months (Figure 1, see Method). 99.45% of reads were human-derived sequences, and the remaining reads were used for taxonomic classification (Figure 1a). Of them, 40 279 (4.34%) were mapped to known microbial references (Table 1). Among the known microbial reads, the top hit detected was EBV with 193 reads (0.47%), followed by Pseudomonas aeruginosa with 189 reads, and Staphylococcus epidermidis with 88 reads. Subsequent in situ hybridization (ISH) confirmed positivity for EBV-encoded small RNAs (EBER) in wound cells (Figure 1b). Serologic testing of this patient revealed past EBV infection. That is, positive for viral capsid antigen (VCA) IgG, and negative for VCA IgM and EBV nuclear antigen (EBNA).

(a) Illustration of metagenomic sequencing and analysis. Reads are first classified into human and non-human sequences. The non-human sequences are mapped to known microbial genomes, revealing presence of EBV; (b) These wound cells are positive for EBER expression by ISH (original magnification ×100)); (c) Atypical lymphocytes infiltration around vessels with destruction of vascular wall (original magnification ×200).
The numbers and percentages of human and microbial reads in the metagenomic sequencing.

Histologic examination revealed an EBV-associated NK/T cell lymphoma
As EBV is often associated with cancers, histology examination was performed. Analysis revealed atypical lymphocytes infiltration around vessels with destruction of vascular wall (angiocentricity and angiodestruction), accompanying extensive coagulative necrosis in the skin, soft tissue, and muscle (Figure 1c). These atypical lymphocytes were medium-sized with irregular nuclear contours and pale cytoplasm. Immunohistochemical analysis showed that the tumor cells were positive for CD3 and T-cell intracellular antigen-1, and negative for CD20 and BCL-2. As a consequence, extranodal NK/T-cell lymphoma was reported as the etiology of the chronic wound.
Classification of type 1 EBV by type-specific proteins
These viral reads were evenly distributed across the entire EBV genome (Figure 2a), implying they were not artifacts of PCR amplification. For classifying EBV Types 1 or 2, the viral reads were mapped to 6 type-differentiating proteins (eg, EBNA-2, EBNA-3A, see Methods). The protein of type 1 EBNA-3A was mapped by 6 reads of nearly all of them showing 100% identity (Figure 2b), while protein type 2 EBNA-3A was only mapped by 4 reads with lower nucleotide identities (81.25%-93.75%) (Supplemental Figure 1). Hence, the virus in the wound sample was classified as type 1 EBV.
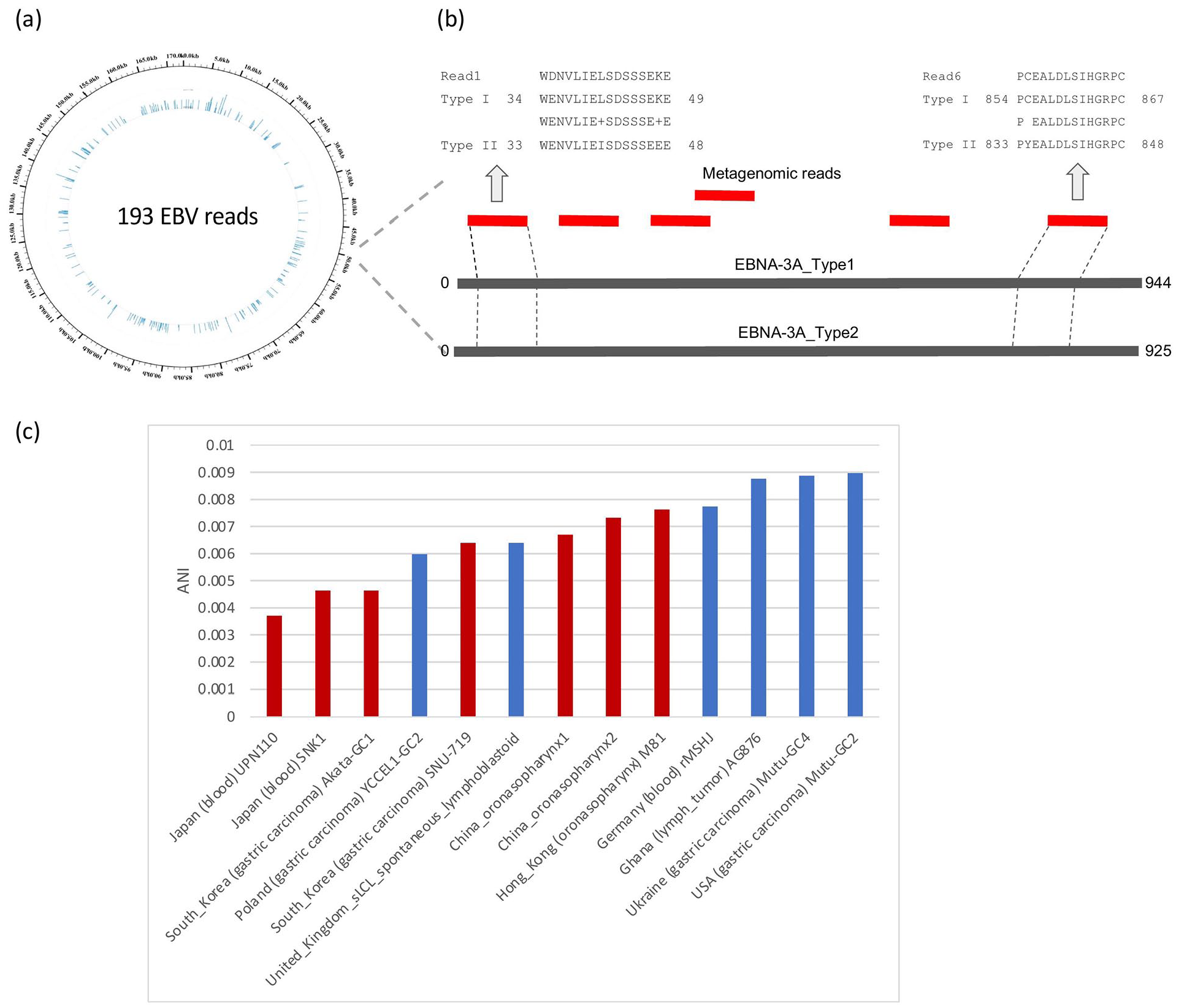
(a) Distribution of 193 EBV reads on the EBV genome; (b) Illustration of 6 reads mapped to the proteins of type one-half EBNA-3A. The alignments of 2 reads are enlarged on the top; (c) Distances measured by the average nucleotide identity of Type 1 EBV strains from different geographical regions.
Similarity of EBV strains shaped by geographical proximity
Thanks to the uniformly-distributed reads across the EBV genome, 69 mutations were detected in the wound strain by mapping reads against the EBV reference (B95-8). We investigated the average nucleotide identity of our strain (B95-8-LT) with respect to other Type 1 EBV strains across a range of geographic locations and tissue types (Figure 2c). The results indicated that the B95-8-LT strain isolated in Taiwan was more similar to those EBV strains identified within Asia (eg, Hong Kong, Japan) than to those outside strains detected and identified outside Asia. Although the AG876 strain (in Ghana) was also originated from a lymph tumor, the genome was dissimilar when compared to our B95-8-LT strain. As a consequence, the similarity of EBV genomes is mainly shaped by geographic proximity rather than tissue types.
Discussion
Using ultra-deep metagenomic sequencing enables researchers to identify viruses and bacteria although human DNA material is highly abundant. In our study, we recovered no less than 193 viral reads belonging to EBV that were uniformly distributed across the genome. Uniform distribution increased the chance of identifying type-specific mutations and comparison of strains from a variety of geographical locations. The strain detected in our study belonged to type 1 EBV and 69 mutations were detected when compared to the EBV reference genome (B95-8).
In vitro data suggests that type 1 EBV is more capable than type 2 EBV to sustain lymphoblastoid cell proliferation. Differences between the 2 types of EBV are also found in the regulatory regions or coding regions of a variety of other genes, including EBNA1, LMP1, and ZTA, as well as other viral proteins which have been identified and play a role in the proliferation of lymphoblastoid cell lines.13,14 Previous studies suggest that the type 1 EBV is most commonly identified in tumors and is responsible for causing acute infectious mononucleosis, while the type 2 virus has been identified in some African Burkitt lymphoma (BL) and some AIDS-associated lymphoma. 13 Hence, detecting the type 1 EBV in this study is in agreement with previous findings and associations.
Epidemiological studies regarding geographical distribution demonstrate that EBV type 1 distributes worldwide while type 2 strains are less common.15-17 Our analysis using EBV genomes from Asia, America, Africa, and Europe revealed that EBV strains form clusters according to their geographical proximity. Analysis using the fragments covered (not complete genome) of the strain detected in this study (Taiwan) reveals higher nucleotide similarity with strains detected and identified in Asia than to those found outside Asia.
We note that initial analysis using incomplete EBV genomes in NCBI failed to reveal the geographical clusters. Therefore, the correctness of phylogenetic analysis of EBV seems heavily affected by the completeness of viral genomes. We ever conducted a phylogenomic analysis by constructing a hybrid genome which replaced the EBV reference with our 193 reads (Supplemental Figure S2). While the phylogeny reconstructed was concordant with geographical clusters implied by ANI (Supplemental Figure S2), the genomic distances measured by the hybrid genome were still untrue. Hence, as most microbial genomes in clinical metagenomic sequencing are incomplete, better distance estimation and/or recalibration methods are necessary for producing accurate phylogeny.
Supplemental Material
sj-pdf-1-evb-10.1177_11769343221110663 – Supplemental material for Metagenomic Sequencing and Histology on a Chronic Wound Identified Epstein-Barr Virus-Associated Lymphoma
Supplemental material, sj-pdf-1-evb-10.1177_11769343221110663 for Metagenomic Sequencing and Histology on a Chronic Wound Identified Epstein-Barr Virus-Associated Lymphoma by Wan-Ting Yang, I Chiang, Chien-Hao Tseng, Chun Cheng, Jyun-Hong Lin, Po-Yu Liu and Yao-Ting Huang in Evolutionary Bioinformatics
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: YTH was supported in part by the Ministry of Science and Technology with grant No. 109-2221-E-194-038-MY3.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Consent for Participation and Publication
Informed consent was obtained from the subject.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.