
Abstract
Statins, a class of cholesterol-lowering medications that inhibit 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase, are commonly administered to treat atherosclerotic cardiovascular disease. Statin use may expand considerably given its potential for treating an array of cholesterol-independent diseases. However, the lack of conclusive evidence supporting these emerging therapeutic uses of statins brings to the fore a number of unanswered questions including uncertainties regarding patient-to-patient variability in response to statins, the most appropriate statin to be used for the desired effect, and the efficacy of statins in treating cholesterol-independent diseases. In this review, the adverse effects, costs, and drug–drug and drug–food interactions associated with statin use are presented. Furthermore, we discuss the pleiotropic effects associated with statins with regard to the onset and progression of autoimmune and inflammatory diseases, cancer, neurodegenerative disorders, strokes, bacterial infections, and human immunodeficiency virus. Understanding these issues will improve the prognosis of patients who are administered statins and potentially expand our ability to treat a wide variety of diseases.
Keywords
Overview of Current Statin Use
Cardiovascular disease (CVD) is the leading cause of worldwide deaths, with mortality rates of approximately 235 per 100,000 inhabitants. 1 In the vast majority of patients, CVD is specially attributed to atherosclerosis. While the development of atherosclerotic CVD (ASCVD) involves a variety of biological processes and behavioral factors, high plasma cholesterol levels are thought to play a primary causative role. 2 Thus, the preferred treatment of hypercholesterolemia-induced cardiovascular-associated diseases, which includes ASCVD, involves the use of statins, which are highly effective in lowering the cholesterol levels. 3
Lovastatin became the first commercially available statin medication in 1987 when it was given the United States Food and Drug Administration (FDA) approval. 4 Since then, the use of statins has proven to be advantageous for the primary prevention of CVD by reducing the risk and preventing the onset of the disease. 5 In addition, statins are often used for secondary prevention as they are effective in slowing disease progression and reducing cardiovascular-associated morbidities and mortalities. In 2013, the American College of Cardiology (ACC) and the American Heart Association (AHA) revised the existing guidelines to reduce the risk of CVD. 3 Since these guidelines significantly expand the population of patients eligible for statin therapy, it is likely that statin use will dramatically increase. 6 An increase in the use of statins is also thought to arise from their cholesterol-independent (pleiotropic) effects, which have implications in a wide variety of disease processes and thus may significantly broaden their therapeutic use. 7 Given that the patient population thought to benefit from the use of statins will likely continue to increase, it is imperative that factors associated with statin use such as patient costs, their adverse effects (AEs), and their interactions with food and other drugs be thoroughly understood. Furthermore, the effectiveness of statins in treating diseases other than ASCVD, such as autoimmune/chronic inflammatory diseases, cancer, neurodegenerative disorders, stroke, and bacterial and viral infections should be clearly established.
This review summarizes the most recent literature regarding traditional statin therapy, their associated pleiotropic effects, and the current and emerging therapeutic applications of statins. Content within this review may be of value for both the medical and research communities in guiding efforts toward optimizing patient care and furthering our understanding of issues pertaining to the use of statin therapy for treating a plethora of disease states.
Current Therapeutic Applications of Statins
Goals of statin therapy
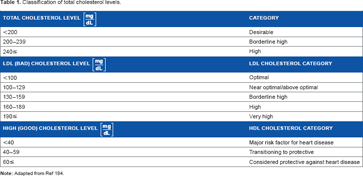
Classification of total cholesterol levels.
LDL and high-density lipoproteins
Lipoproteins are particles that play a major role in transporting lipids, in circulation, such as cholesterol and triglycerides. 9 Lipoproteins are classified primarily according to their density and lipid composition and consist of the following seven major types: chylomicrons, chylomicron remnants, very low-density lipoproteins (VLDL), intermediate-density lipoproteins (IDL), LDL, high-density lipoproteins (HDL), and lipoprotein-(a). Chylomicrons carry dietary triglycerides and esterified and unesterified cholesterol from the intestine to the liver. While both VLDL and IDL are involved in the transport of lipids from the liver to the peripheral tissues, VLDL exports primarily triglycerides, whereas IDL exports both triglycerides and cholesterol. LDL is highly enriched in cholesterol, transporting the majority of cholesterol found in the circulation. HDL transports both cholesterol and phospholipids but reverses the transport of the lipids by carrying them from the peripheral tissues back to the liver where they are recycled and excreted. The physiological function of lipoprotein-(a), an LDL particle, has not yet been defined.
LDL-C is often referred to as bad cholesterol due to its close association with ASCVD, whereas HDL-C, because of its role in reverse cholesterol transport and inverse relationship with cardiovascular incidents, is known as good cholesterol. 2 The causal relationship between LDL-C and ASCVD was first established by using human genetic analyses. 10 Here, patients with familial hypercholesterolemia and elevated LDL-C levels (due to loss-of-function mutations in the LDL receptor) were found to develop early ASCVD. Furthermore, patients expressing various forms of additional members of the LDL pathway, such as protein convertase subtilisin/kexin type 9 (PCSK9), exhibited low LDL-C levels, which corresponded to a reduced risk of developing ASCVD. In addition, pharmacological analyses of the effectiveness of statins have established a link between statin use and significant reductions in coronary events regardless of gender. 11 Thus, use of both genetic analysis and intervention strategies has firmly established the importance of targeting LDL-C via the use of statins as an effective approach for treating ASCVD.
Alternatives to statin use
While statins have proven to be advantageous for lowering the LDL-C levels in the majority of individuals, some individuals fail to respond to treatment (statin resistant) or are prone to developing AEs (statin intolerant). 12 Patient's response to statins varies widely with reductions in LDL-C levels following the administration of statins ranging from 5% to 70%. Patients who are statin resistant do not achieve desired LDL-C target levels even when a high dose of a potent statin is administered. Statin resistance likely arises from a number of mechanisms including polymorphisms in genes involved in cholesterol synthesis and metabolism, such as 3-hydroxy-3-methyl-glutaryl-coenzyme A reductase (HMGCR) and low-density lipoprotein receptor (LDL-R), and those associated with statin pharmacokinetics, such as transporter proteins (eg, ATP-binding cassette sub-family G member 2 [ABCG2] and SCLO1B1). Patients who are statin intolerant typically exhibit sensitivity to statin-induced myopathy and/or liver injury as indicated by increases in liver enzyme activity. In both statin-resistant and statin-intolerant patients, other avenues of treatment must be explored. 3 Alternatives to statin therapy include use of cholesterol absorption inhibitors and bile-acid sequestrants; however, they lack the potency of statins in their ability to lower LDL-C levels. The need for more effective LDL-C-lowering therapies, particularly for treating very high-risk patients, has led to the development of PCSK9 inhibitors. 13 However, their relatively high cost and the limited data surrounding their clinical effectiveness currently prohibits their wide-spread use. Thus, current research and clinical efforts will continue to focus on developing effective alternatives to statins in patient populations where statin therapy is contraindicated.
Additional approaches, including specific dietary recommendations, such as fish and fish oil, and exercise, should be also considered.14,15 For example, patients who altered their diets and engaged in exercise programs of 10 miles of walking or jogging per week showed a more substantial 14%–20% decrease in LDL-C levels as compared to those who only altered their diets. 15 While a number of dietary regimens have been designed to decrease LDL-C levels, their effectiveness varies extensively. The most stringent diets, such as the AHA Step-1 diet, and the Mediterranean diet, only elicit a reduction in LDL-C levels by approximately 5%–9%.16,17 Diets such as the Ornish diet have the ability to produce more impressive results, decreasing serum LDL-C levels by approximately 17%. 18 However, many of these diets may not be realistic in practice as they vary considerably from the typical modern American diet. The benefits reported with diet alone should be contrasted with the benefits of statins as a routine secondary prevention (35% cardiac event reduction). Diet and exercise should be considered vital to the reduction of high cholesterol and prevention of cardiac events. However, diet and exercise may not be able to lower LDL levels to less than 100 mg/dL. In these instances, a pharmacologic statin therapy should be maintained with the continuation of diet and exercise.
Statin mechanisms of action
The current therapeutic goal of statins involves the following mechanisms
19
(as illustrated in Fig. 1). Statins inhibit HMGCR in hepatocytes. HMGCR is the rate-limiting enzyme of the hepatic cholesterol synthetic pathway and converts 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) to melvalonic acid–-a precursor in the de novo cholesterol biosynthetic pathway. Statins reversibly compete with the endogenous substrate, HMG-CoA, for the active site within the reductase. Binding of the statin to the active site of the enzyme confers a conformational change that attenuates the enzyme's function. Acting as competitive antagonists, the higher affinity statins bind to the active site of the enzyme, thereby preventing binding of the lower affinity endogenous substrate, HMG-CoA (Fig. 1A). The resultant conformational change and inhibition of HMGCR decreases cholesterol production and reduces the intracellular cholesterol stores of the hepatocytes (Fig. 1B). In an effort to maintain homeostasis and counteract this decline in intracellular cholesterol, a protease is triggered to cleave the sterol regulatory element-binding protein (SREBP) from its protein precursor in the endoplasmic reticulum. The unbound SREBP is subsequently translocated to the nucleus (Fig. 1C). In the nucleus, SREBP binds the sterol regulatory element (SRE) located within the promoter elements of the gene encoding LDL-R (Fig. 1D). Transcription of the LDL-R gene is enhanced resulting in increased expression of LDL-R mRNA and increased synthesis of LDL-R protein (Fig. 1E). The hepatic LDL-R protein undergoes maturation and constitutive exocytosis intended for hepatocyte surfaces (Fig. 1F). Free LDL-C binds to the newly synthesized LDL-R leading to endocytosis and subsequent lysosomal degradation of LDL-C within the hepatocyte (Fig. 1G). The internalization of LDL-C increases intracellular cholesterol levels and promotes a return to homeostatic levels of LDL-C (Fig. 1H). The ultimate effect of this series of events is a reduction in circulating LDL-C levels–-elicited in large part by the increase in hepatic LDL-R cell surface density (Fig. 1I).
Statin mechanism of action. (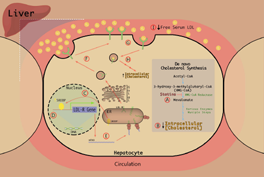
Use of statins for reducing the risk of ASCVD
Recently, the ACC and AHA approved the revision of existing guidelines designed to reduce the risk of ASCVD that were based on evidence generated from a series of randomized controlled trials.
3
While previous recommendations focused on specific target levels of LDL-C, the new recommendations defined patient groups who would most likely benefit from moderate- or high-intensity statin therapy. These groups have been identified as follows:
clinical ASCVD, ie, acute coronary syndromes, or a history of myocardial infarction, stable or unstable angina, coronary or other arterial revascularization, stroke, transient ischemic attack, or peripheral arterial disease of atherosclerotic origin; primary elevations of LDL-C ≥190 mg/dL; age 40–75 years with diabetes and LDL-C 70–189 mg/dL; age 40–75 years with LDL-C 70–189 mg/dL and estimated 10-year ASCVD risk ≥7.5%.
As these guidelines are clinically implemented, it is predicted that 12.8 million additional individuals will now qualify for statin therapy. 6 As statin use continues to increase, a concern is raised that patients, as well as physicians, may become over-reliant on pharmacological methods of controlling high cholesterol levels and disregard approaches such as lifestyle changes that are beneficial for not only improving cardiovascular health 20 but also overall well-being. Lifestyle changes such as the elimination of tobacco products, dietary modifications, weight management, exercise, and yoga have been shown to be effective in reducing the risk of CVD. 21 Thus, it is recommended that patients who qualify for statin therapy would benefit the most from intervention strategies that would include statin therapy coupled with a healthy lifestyle.22,23
Potential AEs associated with statin use
While statins, in general, are well tolerated, AEs are reported and include muscle pain and damage, increased blood glucose levels, which may contribute to type 2 diabetes mellitus (T2DM), hepatotoxicity, digestive problems, cognitive effects, and the development of rashes or flushing. 24 Of these AEs, those impacting muscle, blood glucose levels, and liver function are thought to be the most clinically relevant.
Muscle pain and damage
Statin-associated muscle symptoms (SAMS) are the most commonly reported AEs affecting up to 30% of the patient population. 25 SAMS typically present as muscle pain, soreness, aching, stiffness, etc, and are often the source of statin nonadherence or discontinuation. Clinically, these symptoms can be defined as myopathy (muscle weakness), myositis (muscle inflammation), myonecrosis (elevated muscle enzyme levels), rhabdomyolysis (severe myonecrosis with myoglobinuria or acute renal failure) and myalgia (unexplained muscle discomfort which encompasses muscle aches, soreness, etc). Mild myalgia occurs in 5%–10% of statin users annually and is often intermittent. Life-threatening rhabdomyolysis occurs in only 0.001%–0.005% of patients or 1–5 of every 1000 statin users annually. 24 Concerns related to the risk of rhabdomyolysis have led to the removal of cervastatin from the market and recommendations that patients who are administered high doses of simvastatin (~80 mg) be closely monitored.26,27 Statins are thought to adversely impact muscle tissue by altering mitochondrial function and cellular energy utilization as well as depleting coenzyme Q10 levels. 25 Patients who are at high risk for developing statin-induced muscle symptoms are often those with other comorbidities or who are coadministered other drugs as described in the following section.
Type 2 diabetes
The question of whether statin use significantly raises blood glucose levels and contributes to the development of T2DM is complicated by the fact that high LDL-C, ASCVD, and T2DM share common risk factors. 28 Like ASCVD, behavioral risk factors of T2DM include a lifestyle devoid of physical activity, a high calorie diet saturated with high trans-fat foods, cigarette smoking, and overweight/ obesity. Other contributing risk factors are socioeconomic and include race/ethnicity, culture, and geographical location. 29 Thus, the likely use of statins by patients with T2DM may be deleterious with respect to their T2DM-related conditions.
The diabetogenic effects of statins are thought to arise from several mechanisms that converge on glucose regulation and pancreatic beta cells,30,31 including inhibition of isoprenoid synthesis and subsequent inhibition of glucose uptake by beta cells, increased uptake of LDL leading to glucokinase inhibition (hence blocking glucose conversion to pyruvate), and cytokine-induced overproduction of nitric oxide (NO) leading to beta cell apoptosis. In addition, statins suppress ubiquinone and ATP synthesis. These mechanisms may converge or they may in fact be mutually exclusive, yet the ultimate effect rendered is one that quells insulin release from beta cells.
A thorough review of the literature reveals the following three key themes that are important for understanding the relationship that may exist between statin therapy and T2DM.
Because of the mechanisms discussed earlier, statins may raise blood sugar levels. Numerous population-based studies have consistently reported that compared to patients in the placebo group, a greater number of patients receiving statin therapy were subsequently diagnosed with T2DM.30–33 While there have been incident cases of T2DM in longitudinal, randomized clinical trials within a statin therapy group, there is less often a significant difference in these incident cases occurring in the treatment group versus the control group. The findings of one study revealed that 3% of the treatment group (rosuvastatin) developed T2DM while only 2.4% of the placebo group developed the disease (P = 0.01).
34
However, those patients of the treatment group also had a significant reduction in the risk of heart attack (54%), 48% reduced risk of stroke, and 20% lower risk of mortality. CVD risk is increased twofold to fourfold in patients diagnosed with T2DM.
35
In T2DM patients with normal LDL levels, statins may have a major role in preventing the long-term negative health effects often associated with chronic T2DM. As in the first case, the long-term benefits of a therapeutic statin regimen may outweigh any undesirable effects. Statins may have a diabetogenic potential through the various mechanisms discussed earlier. While new-onset T2DM is observed with all statins,
33
and associations coupled from those observations, a causal relationship cannot be implicated as numerous patients of the control group (receiving a placebo) also in fact developed T2DM. However, it has become widely accepted that this relationship is dependent on statin type and dose.28,36
It is thought that “statin therapy is associated with a slightly increased risk of developing of diabetes.” 35 However, the risk is low, and when compared to the reduction in coronary events, the benefits of statin therapy outweigh the risk of developing diabetes.
Liver function
Statin-induced hepatotoxicity is relatively rare, and approximately 3% of patients who take statins develop elevated transaminase levels, a marker of liver injury. 37 The increases in transaminase levels are typically temporary and thought to be due to decreased cholesterol levels, increased membrane permeability, and leakage of liver enzymes. In a recent study, 22 out of 1188 cases of drug-induced liver injury were attributed to statin use. 38 Here, statin-induced liver injury was primarily of mild-to-moderate severity and reversible and was typically observed after months or years of statin use. Thus, as endorsed by the FDA, currently marketed statins are associated with a very low risk of serious liver injury.
Patient-to-patient variability
Differences in response to statin drugs among different patient populations.

The wide variability in statin plasma levels and statin-induced responses in patients following a given statin dose is linked to polymorphisms of genes involved in the import, export, and metabolism of statins. 41 Of particular interest are cytochrome P450s (cytochrome P4502C9 [CYP2C9] and cytochrome 3A4 [CYP3A4]) and drug transporters (ie, solute carrier organic anion transporter family member 1B1 [SLCO1B1], which facilitates hepatic statin uptake and the ABCG2 export pump). While CYP2C9 plays a key role in metabolizing fluvastatin, CYP3A4 is of more importance in metabolizing simvastatin and lovastatin. Patients expressing various forms of CYP2C9, with reduced enzyme activity, are more responsive to the LDL-C-lowering effects of fluvastatin. 42 Similarly, patients expressing forms of CYP3A4 with reduced enzyme activity are more responsive to the LDL-C-lowering effects of simvastatin, atorvastatin, and lovastatin.43,44 Furthermore, patients who harbor allelic variations of both CYP2C9 and hepatic transporters (ie, OATP1B1) may be at high risk for developing fluvastatin-induced myotoxicity. 45 Finally, in patients expressing a SLCO1B1 variant that is defective in hepatic statin uptake, high statin plasma levels and elevated risks of statin-induced myopathy have been observed. 46 In fact, a recent study reported that polymorphisms in SLCO1B1 and ABCG2, which correspond to impaired transporter function, accounted for 90% of the patient variability of rosuvastatin plasma levels. 47 Taken together, these studies are indicative of the significant advances made in our understanding of how polymorphisms in drug-metabolizing enzymes and transporters contribute to variations in patient's response to statins. As a result, the decision-making process involved in weighing the benefits and risks associated with statin use will likely continue to improve.
Statin use is associated with drug–food and drug–drug interactions
Statins are involved in a variety of interactions with food and other drugs and typically involve inhibition of CYP2C9, CYP3A4, and transporters. 48 Statin interactions may be divided into the following two groups based on their metabolic pathways: (1) those metabolized by CYPs; CYP3A4 (simvastatin, lovastatin, and atorvastatin), and CYP2C9 (fluvastatin) and (2) those not metabolized by CYPs. While both simvastatin and lovastatin are highly involved in drug– drug interactions, for simplicity, the discussion below will be limited to interactions involving simvastatin.
Statins and grapefruit
The most commonly reported food–drug interaction involves grapefruit, which contains furanocoumarins, potent CYP3A4 inhibitors. 49 Thus, ingestion of grapefruit can impair a patient's ability to metabolize drugs like simvastatin, resulting in a significant increase in plasma simvastatin levels. For example, the ingestion of grapefruit juice (200 mL) can increase simvastatin plasma levels by more than threefold. 50 In some cases, the statin/grapefruit interaction can result in rhabdomyolysis. 51 At this time, however, the prevalence between severe, statin-induced AEs and the consumption of grapefruit in the general population is difficult to ascertain.
Statins and other drugs
Drug–drug interactions involving simvastatin have been reported in patients concomitantly administered drugs that are potent inhibitors of CYP3A4, such as itraconazole, ketoconazole, amiodarone, cyclosporine, ritonavir, and indinavir. 48 Coadministration of simvastatin with antifungals (itraconazole or ketoconazole) can result in rhabdomyolysis and acute renal failure.52,53 Similar AEs may also arise upon coadministration of simvastatin with amiodarone, an antiarrhythmic agent.54,55 In fact, the FDA recommends that in patients taking amiodarone, the dose of simvastatin should not exceed 20 mg/day. Concerns are also expressed in patients treated with simvastatin and cyclosporine, an immunosuppressant.56,57 Here, the high prevalence of rhabdomyolysis and acute kidney injury have triggered warnings that the coadministration of these two drugs should be avoided. The AEs associated with cyclosporine use involve not only its inhibition of CYP3A4 but also a number of influx and efflux transporters. 58 Finally, rhabdomyolysis has been reported when patients were cotreated with simvastatin and HIV protease inhibitors such as ritonavir and indinavir. 40 Because of these reported events, the FDA has recommended labeling changes for several statins (simvastatin, lovastatin, atorvastatin, and rosuvastatin) to include contraindications, cautions against, or limited doses to reflect the risks for interactions between these statins and HIV and HCV protease inhibitors.
The costs associated with the use of statins and other cholesterol-lowering drugs
Costs associated with the use of statins and other cholesterol-lowering drugs.
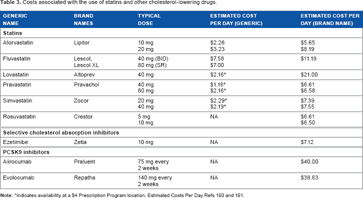
Indicates availability at a $4 Prescription Program location. Estimated Costs Per Day Refs 190 and 191.
Comparison of individual statins
While statins are often thought of as a singular entity with similar pharmacological effects, they differ significantly with respect to their chemical properties, efficacy, safety, and the frequency and type of side effects experienced. Extensive meta-analyses and randomized trials have addressed this issue by comparing a variety of different statins. Combining the results of these studies, along with the cost of each drug as shown in Table 3, demonstrates which statin may be the best choice for a particular patient.
The STELLAR study found that atorvastatin at doses between 10 and 80 mg had the highest number of treatment discontinuations due to AEs, followed by rosuvastatin, simvastatin, and pravastatin (although there was no 80 mg group for pravastatin). 60 These results coincide with the findings of a meta-analysis that simvastatin and pravastatin appear to be the most tolerable and safest of the tested statins. 61 The STELLAR study also found that rosuvastatin had the highest average reduction in LDL-C from baseline at 51.06% (albeit no reported data at an 80 mg dose), followed by atorvastatin at 44.58%, simvastatin at 36.98%, and pravastatin at 24.7%. 60
An additional meta-analysis of randomized trials, the VOYAGER study, consisting of 32,258 patients, investigated various statin therapies and compared their impact on the reduction in LDL-C and triglycerides in patients with hypertriglyceridemia–-a condition associated with an increased risk of CVD. Here, rosuvastatin was found to reduce LDL-C on average, by 52% at a lower dose (40 mg) than both atorvastatin and simvastatin at a higher dose (80 mg), of which showed a mean reduction of 49% and 43%, respectively. 62
Narrowing the focus to specific potential AEs, interesting findings surrounding the development of T2DM and SAMS, also referred to as muscle AEs, when comparing different statins are revealed. An analysis involving 246,955 individuals reported that the only AEs with higher odds ratios (OR, 1.51) occurring with statin use compared to a control were elevated transaminase levels and the development of T2DM (OR, 1.09). 61 In terms of individual statins, participants were more likely to develop T2DM when taking atorvastatin (OR, 1.17), followed by pravastatin and rosuvastatin (OR, 1.16), simvastatin (OR, 1.10), and lovastatin (OR, 0.98). Fluvastatin did not have a reported OR for T2DM; however, in terms of transaminase elevation, there was a higher odds (OR, 5.18) of occurrence, followed by atorvastatin (OR, 2.55), lovastatin (OR, 2.03), rosuvastatin (OR, 1.59), simvastatin (OR, 1.16), and pravastatin (OR, 1.00).
In addition, a relationship between the potency of statins (as defined by the magnitude by which they reduce LDL-C levels) and reported AEs has been identified in an analysis representing 147,789 cases. 63 Here, symptoms relating to rhabdomyolysis and less severe symptoms (ie, myalgia or joint) were reviewed. Statins with the highest potency per milligram appeared to have the highest AE relative risk. Thus, in a ranking whereby rosuvastatin was designated as 100% relative risk, the less potent statins were ranked as follows: atorvastatin (55%). simvastatin (26%). pravastatin (17%). lovastatin (7.5%). fluvastatin (% not reported). An exception appears to be fluvastatin, a low potent statin with a relatively high relative risk of reported AEs (74%).
Ascertaining the best statin to be used for a particular patient is difficult because of factors associated with the patient (the above mentioned patient-to-patient variabilities in comorbidities, genetic polymorphisms, coexposure to other drugs, etc) and those associated with the individual drug properties (ie, lipophilicity, bioavailability, efficacy, potency, and the prevalence of certain side effects). Pravastatin does not appear to exert an LDL-lowering effect to the extent imposed by the other statins, as reported in the STELLAR and additional studies.60–62 However, it appears to be one of the safest and cheapest options available, followed closely in terms of safety and cost by simvastatin, which typically has a low monthly generic copayment. While simvastatin appears to be the best choice in terms of cost and safety, atorvastatin and rosuvastatin may be more effective with respect to their ability to reduce cholesterol levels but harbor a higher probability of exerting an AE and are also more expensive. Thus, it can be seen that many factors need to be taken into consideration when deciding which statin to administer; it is not a one-size-fits-all therapy, and instead, the therapy should be tailored to the patient in order to accommodate for the aforementioned differences yielding an optimal therapy.
It would be remiss to omit studies reporting findings that vary significantly from other patient populations, globally. In particular, two European studies, the PRIMO survey and the STOMP study, retrospectively found that patients taking simvastatin, atorvastatin, or lovastatin may be at highest risk of developing SAMS (18.2, 9.4%–14.9%).64,65 This variability may be due to the hydrophilic properties of fluvastatin and pravastatin, which may limit their muscle penetration. 64 However, we note that fluvastatin's low association with SAMS supports findings in previous studies that use of lower potency statins may result in fewer AEs. It is evident that further research should be conducted in specific patient populations, with tightly controlled variables. Retrospective data can be skewed by recall bias; therefore, prospective data should be obtained through closely monitored randomized control trials in order to compare specific AEs (such as SAMS) that may result from the use of various statins in defined patient populations.
Summary of statin use and ASCVD
Over the past 30 years, statins have been used as cholesterol-lowering medications. 4 Up to this point, this review has discussed the intended mechanisms of action associated with statins, drug–food interactions, and the use of statins as therapeutic agents for treating patients with ASCVD or those at risk of developing ASCVD. The remainder of this review examines the pleiotropic effects associated with statins and their potential role in abrogating the pathophysiological processes involved in other disease states.
Emerging Therapeutic Applications of Statins
Therapeutic expansion of statin use
Recently, the scope of statin therapy has expanded with the emergence of evidence suggesting that due to pleiotropic effects of statins that are not directly associated with their regulation of cholesterol levels, they may prove to be beneficial for treating a number of diseases.7,6 6 The pleiotropic effects associated with statins that may impact disease pathophysiology include their modulation of immune responses, their enhancement of anti-inflammatory processes, and their alterations of signaling pathways that involve cholesterol intermediates. To date, the multitude of diseases linked to the pleiotropic effects of statins include multiple sclerosis (MS), inflammatory bowel diseases (IBDs), rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), chronic obstructive pulmonary disease (COPD), cancer, strokes, Parkinson's and Alzheimer's diseases, bacterial infections, and HIV. The following section provides an overview of our current understanding of how statins impact the onset or progression of these diseases.
Mechanisms by which statins exert their pleiotropic effects
The pleiotropic effects of statins were first reported in 1995 when it was observed that the benefits of pravastatin extended beyond those attributed to the expected lipid-lowering effects of statin therapy. 67 68 Further investigation revealed that statins possessed novel cholesterol-independent effects, which included stabilizing atherosclerotic plaques, enhancing endothelial function, modulating immune responses, decreasing oxidative stress/inflammation, and inhibiting the thrombotic response. 7 6 6 While statins may be able to modulate the pathophysiology of a variety of disease states, their effectiveness is currently under considerable debate due to the diversity of the involved molecular mechanisms, the wide range of statins’ pharmacokinetic properties, and issues surrounding patient-to-patient variability.
Statins are thought to impact disease processes via mevalonate-dependent and -independent mechanisms.7,69–72 Using a variety of cell types including immune, 69 tumor, 71 and endothelial 72 cells, statins have been shown to alter fundamental cell processes by impinging on well-conserved, key signaling molecules. In addition to the mechanisms described below, those specific to the disease states under discussion are outlined in the following sections.
The mevalonate-dependent mechanisms of statins
The mevalonate-dependent mechanisms arise from statin inhibition of HMGCR activity, cholesterol biosynthesis, and protein isoprenylation.69–72 The isoprenylation of proteins, a posttranslational modification, is required for membrane-associated proteins to form covalent attachments, maintain appropriate subcellular localization, and engage in intracellular trafficking. Key isoprenylated proteins are Ras, Rho, and Rac–-critical GTPases that regulate a variety of biological processes involved in determining cell proliferation, fate, and morphology.
The mevalonate-independent actions of statins
The mevalonate-independent effects of statins involve their inhibition of a variety of signal molecules including lymphocyte function-associated antigen 1 (LFA-1) and intercellular adhesion molecule 1 (ICAM-1). 69 , 70 , 72 LFA-1 modulates leukocyte trafficking and T cell activation, whereas ICAM-1 sustains leukocyte adhesion and facilitates migration. Other mevalonate-independent actions of statins include their inhibition of signal transduction molecules (v-akt murine thymoma viral oncogene homolog [AKT], extracellular signal-regulated kinase [ERK]/mitogen-activated protein kinases [MAPK] and janus kinase (JAK)/signal transducer and activator of transcription [STAT]) and transcription factors involved in mediating proinflammatory responses (nuclear factor-kappa B [NF-κB], activator protein 1 [AP1], histone deacetylase [HDAC], STAT1, STAT3, and STAT4). 69 , 70 In addition, statins activate anti-inflammatory responses by enhancing the activity of transcription factors such as Kruppel-like factor 2 (KLF2) and peroxisome proliferator-activated receptors α and β, which suppress NF-κB and AP1 gene activation.
The extent to which the mevalonate-dependent pathway versus mevalonate-independent pathway contributes to the pleotropic effects of statins has undergone increased scrutiny.69,70 Early interest in the mevalonate-independent pathway was stimulated by the observation that statins are inhibitors of LFA-1 and block its ability to interact with ICAM-1 and thereby inhibit T-cell migration. 73 , 74 However, since excess mevalonate can reverse a statin's ability to inhibit T cell proliferation and function, the importance of statin inhibition of LFA-1 is questionable. 75 , 76 Thus, the extent to which mevalonate-dependent pathway versus mevalonate-independent pathway mediates the pleiotropic and anti-inflammatory actions of statins requires further investigation.
Stains, NO signaling, and cell fate decisions
Importantly, statins also impact vascular and immune functions via altered NO signaling leading to improved vascular function, inhibition of leukocyte chemotaxis, and downregulation of leukocyte adhesion and migration at the vascular wall.69,72 Statin modulation of NO bioavailability involves the following three major mechanisms: direct activation of endothelial nitric oxide synthase (eNOS) activity via activation of protein kinases (AMP-activated protein kinase [AMPK], AKT, and protein kinase A [PKA]), increased eNOS expression via reduction in the Rho/Rho-associated protein kinase (ROCK) pathway, and stabilization of eNOS mRNA and reestablishment of eNOS coupling via upregulation of guanosine triphosphate cyclohydrolase (GTPCH). 77
The current interest in the pleiotropic effects of statins has focused on their ability to modulate cell fate decisions, in particular the proliferation and apoptosis of tumor cells 71 and the differentiation and proliferation of T cells.69,70 Statins inhibit cell proliferation by inhibiting Ras and Rho proteins and stabilizing the levels of cell cycle proteins (p21 and p27). 71 Furthermore, statins induce apoptosis by upregulating proapoptotic proteins (ie, Bax) and downregulating antiapoptotic proteins (ie, Bcl-2). With respect to T cells, statins block T cell activation, migration, infiltration of the target organ, differentiation and proliferation, and the ability to secrete proinflammatory cytokines.69,70 Given that the improper type 17 helper cells (TH17)/regulatory T cells (TREG) balance contributes to autoimmune and inflammatory diseases, increasing efforts are ongoing to devise therapeutic strategies to restore this balance. 78
Do statins inhibit the onset or progression of autoimmune and chronic inflammatory diseases?
In many chronic disease states, the inflammatory response is absconded from its protective role (eliminating a disease stimuli) to a deleterious role of damaging tissue and causing organ failure.78,79 In some diseases, such as MS, the protective versus deleterious switch occurs when self-recognizing antibodies and autoimmunity develop. In other diseases, like IBD, it occurs when loss of immune tolerance and tissue homeostasis are met with an overly aggressive immune response. 76 The immune response triggered by these initial events has been frequently described as dysfunctional or exaggerated and results in chronic and often episodic inflammatory conditions. Regardless of the initial pathological events, the progression of autoimmune/chronic inflammatory diseases typically involves an increase in the severity and frequency of inflammatory episodes (or disease flares), resulting in persistent exposure to the inflammatory milieu causing necrosis and extensive tissue damage. In the majority of patients, an overabundance of cells that exert proinflammatory responses (eg, TH1, TH2, and TH17) and a lack of those mediating anti-inflammatory responses (TREG) are observed. 78 Thus, the ability of statins to alter this imbalance of TH1, TH2, and TH17 cell populations versus TREG cell populations is of particular interest. 74
Statins and MS
The pathology of MS involves TH1/TH17 and cytotoxic CD8 T cells, which contribute to proinflammatory conditions and demyelination of axons in the central nervous system (CNS). 80 As the disease progresses, the neurons degenerate, resulting in the neurological decline associated with MS. The first clinical manifestation, clinically isolated syndrome, may or may not develop into MS. 81 Once diagnosed, MS typically presents as one of the following four types: primary progressive, secondary progressive, relapsing remitting (RRMS), and progressive. RRMS, the most common clinical presentation, involving approximately 80% of all cases, often develops into nonrelapsing secondary progressive MS. In animal models of MS, statins inhibit myelin antigen presentation, block the activation and differentiation of T cells, and reduce the recruitment of leukocytes. 80 The neuroprotective effect of statins may also involve their ability to enhance oligodendrocyte differentiation, reduce oxidative damage, improve vascular function by regulation of NO production, inhibit coagulation, promote angiogenesis, and modulate the peripheral inflammatory response.
In key randomized clinical trials, statin treatment was most beneficial to patients with secondary progressive MS reducing the rate of atrophy by as much as 43%. 82 In patients with RRMS, however, conflicting results have been reported, which is attributed to either the statin dose or complications that may have arisen from their cotreatment with conventional anti-inflammatory agents, either interferon β (IFNβ) or methylprednisone.82–87 As a result, uncertainty exists as to whether or not the coadministration of statins with either IFNβ or methylprednisone is therapeutically beneficial to patients with RRMS. A recent meta-analysis of eight randomized clinical trials has reviewed the effectiveness of statin therapy (simvastatin or atorvastatin) in MS patients. 88 With respect to RRMS, statin therapy used as either a monotherapy or in combination with IFNβ was not found to be therapeutically beneficial. With respect to clinically isolated syndrome, statins, administered as a monotherapy, were not convincingly beneficial. However, with respect to secondary progressive MS, some benefit may be achieved from the use of statins.
Statins and IBD
IBD, composed of Crohn's disease and ulcerative colitis, involves a variety of genetic and environmental factors, a dysregulated immune response mounted against the intestinal microbiota and chronic intestinal inflammation. 89 The majority of studies performed using IBD-relevant animal models have found that statin administration alleviates most of the observed inflammatory symptoms. 90 In patient populations, statin exposure has been associated with a significant decrease in IBD onset, particularly in older (>60 years of age) patients. 91 Furthermore, in patients diagnosed with IBD, statin use was associated with a reduced use of oral glucocorticoids, implying that statins reduced the severity of their disease conditions. 92 Other studies have found that IBD patients who were treated daily with statins (atorvastatin) had significantly reduced plasma levels of markers of systemic inflammation (ie, C-reactive protein).93,94
Statins and RA
RA is characterized by chronic inflammation of the joints, synovial hyperplasia, bone destruction, joint deformity, and systemic inflammation. 95 Patients with RA are at an increased risk of mortality, as much as 50%, due primarily to increases in CVDs.96,97 A recent meta-analysis involving 992 RA patients demonstrated that statin use significantly decreased serum levels of inflammatory markers. 98 Furthermore, patients taking statins have a reduced incidence of RA, 99 and in those taking simvastatin and atorvastatin, a reduction in markers indicative of both RA and CVD has been reported.100,101 Despite these promising early studies, further investigation is needed to supplement the notion of a statin as an effective therapy to treat RA.
Statins and SLE
SLE is an autoimmune disease that involves aberrant B cell and T cell signaling and dysregulated apoptosis.102,103 The cellular debris that accumulates provides a source of autoantigens, which contributes to chronic inflammation, formation of immune complexes in specific organs, and organ failure. SLE patients are at high risk for developing CVD, which is thought to arise from a shared pathogenesis. 104 However, randomized controlled trials have failed to demonstrate an effect of statin therapy on SLE disease activity, despite observed reductions in serum C-reactive protein levels. 105 Other studies indicate that in SLE patients with hyperlipidemia, statins may reduce the risk of mortality. 106 In some patients, however, the development of autoimmune reactions has been noted, particularly when either simvastatin or atorvastatin was administered.107,108 Autoimmune reactions involving these second-generation statins may arise from their proapoptotic activities as well as a statin-induced shift from TH1 to TH2 immune responses, enhanced B cell reactivity, and the production of pathogenic autoantibodies. Thus, use of statins for treating SLE holds promise, but the possible development of autoimmune reactions in SLE patients requires further scrutiny.
Statins and COPD
The pathology of COPD, an irreversible and progressive obstruction of airflow, also involves an abnormal inflammatory response. 109 While statins have been considered for use in treating COPD, current guidelines do not recommend the use of statins to prevent acute disease exacerbations. 110 The basis for these guidelines stems from the discrepancy between results obtained from retrospective cohort studies indicating a beneficial effect of statins in COPD patients and those from randomized clinical studies, which have failed to confirm these findings.109–112
Do statins inhibit the onset and/or progression of cancer?
The idea that statins may exert anticancer effects arose in part from reports that statin use was associated with a significant reduction in colorectal cancer incidences 113 and a reduction in mortality associated with 13 different cancer types. 114 However, as discussed in more detail below, epidemiological and clinical studies have thus far failed to consistently substantiate claims that statins are effective chemopreventive/chemotherapeutic agents but instead have underscored the complexities associated with the development and treatment of human cancers. Our analysis of statins and cancer will be limited to cancers of the colon, breast, and prostate as these are among the top five most commonly diagnosed cancers worldwide. 115 Furthermore, the analyses of the impact of statins on the onset, progression, and cancer-specific mortality of colorectal, breast, and prostate cancers are relatively extensive. The steroid responsive cancers (breast and prostate) are of particular interest as both the disease etiology and therapy hinge on the activities of estrogen and androgen, respectively, which may be modulated by statins within the breast or prostate tumor cell. By comparing and contrasting the impact of statins on the onset and progression of these three cancer types, we aspire to identify commonalities and major mechanistic pathways that may be relevant to the majority of human cancers.
Statins as potential chemopreventive agents
The question of whether statin use prevents the onset of colorectal, breast, or prostate cancers is confounded by the contradictory nature of the reports from observational studies. For example, the impact of statin use on colorectal cancer incidence has been widely studied for more than a decade. 116 Despite this, a recent meta-analysis representing more than 8 million patients and employing a variety of methodological approaches has failed to convincingly and consistently demonstrate that long-term use of statins prevents the onset of colorectal cancer. Perhaps more troubling are reports indicating that statin use may increase the risk of colorectal cancer. 117 Similar contradictory findings have been reported with respect to the impact of statin use on breast cancer incidences where an analysis of more than 77,000 women found that the risk of developing breast cancer was not associated with statin use regardless of use duration. 118 Others report that extended statin use is associated with an increased risk of developing breast cancer. 119 With respect to prostate cancer, the evidence pertaining to statin use is similarly contradictory. Some studies report that statin use is not associated with the risk of developing prostate cancer.120,121 Others report that statins may decrease the risk of developing low-grade, early-stage prostate cancer but not advanced stage prostate cancer. 122 In contrast, yet others report that statin use was not associated with a decrease in overall risk of prostate cancer but rather a decrease in the risk of developing advanced prostate cancer.123,124
In addition to preventing the onset of cancer, statins have potential for inhibiting cancer metastasis and improving patient survival. Thus far, reports from phase II/III clinical trials indicate that statin use does not improve progression-free survival of patients who have been diagnosed with metastatic colorectal tumors.125,126 With respect to breast cancer, some studies have found no association between statin use and breast cancer mortality, 127 while others suggest that statins may decrease breast cancer mortality128,129 particularly, if the most lipophilic statin, simvastatin, had been administered. 130 However, reports focused on prostate cancer are more optimistic with some studies suggesting that statins attenuate prostate cancer progression131,132 and mortality.133–135
A number of issues may contribute to the contradictions surrounding the question of whether statins are effective anticancer agents. First, is the timing of the events associated with the development of a tumor relative to the duration of statin use. The majority of human tumors are likely caused by a series of mutational events that sequentially occur over a span of 20–30 years depending on the affected genes and involved cellular processes. 136 Thus, a statin may effectively modulate the relatively modest changes in cellular processes that form within a cell of an emerging tumor (ie, within years 1–10 of tumor development) but may lack potency in overcoming the plethora of metabolic and cellular processes that exist within a fully developed tumor at the end of this 20-to 30-year period. Second, the role of cholesterol in tumor development is undefined and may present an indication bias when statins are used to treat high cholesterol. 137 The available evidence indicates that high blood cholesterol (an indicator for the administration of statins) may be associated with a decrease in colorectal cancer incidence 137 but an increase in prostate cancer incidence. 138 Third, cancer is a highly heterogeneous disease composed of distinct subtypes that likely determine a patient's tumor development and response to a particular drug therapy or chemopreventive agent. 139
It is possible that only a specific tumor subtype can respond to the anticancer effects of statins. With respect to colorectal tumors, molecular subtyping aligns with tumor progression involving activation of oncogenes (ie, Kirsten rat sarcoma viral oncogene homolog [KRAS]) and inactivation of tumor suppressor (ie, adenomatous polyposis coli [APC]) and DNA repair genes. In IBD patients, tumor development is also influenced by the chronic inflammatory conditions within the colon. 140 With respect to breast cancer, molecular subtyping relies on tumor expression of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor 2 (HER2). Ongoing molecular characterization of prostate cancer includes aberrant androgen receptor signaling, as well as genes such as ETS protooncogene (ETS), KRAS, phosphatidylinositol 3-inositol (PI3K)/AKT, phosphatase and tensin homolog (PTEN), and tumor suppressor p53 (TP53). 141 Studies examining the impact of statin use on breast cancer subtypes indicate that statins may increase 142 or decrease 143 the risk of developing ER-negative breast cancers or may increase the risk of developing PR-negative breast cancer. 144 However, others report that statin use is not associated with incidence regardless of the breast cancer subtype that has developed. 145 Whether or not statins impact specific subtypes of colorectal and prostate tumors is yet to be determined.
Putative chemopreventive mechanisms of statins
Efforts focused on identifying patients most likely to respond to the anticancer effects of statins have uncovered a few important underlying genetic and mechanistic events. For example, patients who harbor a form of HMGCR that is less effective at binding statins appear to be less responsive to a statin's ability to reduce the onset of colorectal cancer. 146 Furthermore, in patients undergoing androgen deprivation therapy, the ability of statins to delay prostate tumor progression appeared to involve statin interaction with solute carrier organic anion transporter family member 2B1 (SLCO2B1). 131 It is proposed that by interacting with the SLCO2B1 transport protein, statins competitively displace androgens, thereby preventing their entry into the tumor cell and inhibiting their protumorigenic activities. Similar events may occur in breast cancer patients and thereby alter their response to antiestrogens, since estrone 3-sulfate, a component of the estrogen pool, is also subject to SLCO2B1 transport. 147
Attempts to identify appropriate biomarkers to assess the chemotherapeutic effectiveness of statins include a clinical trial, wherein a relatively high statin dose (ie, 80 mg/day) resulted in a decreased tumor expression of Ki67, a marker of proliferation, and a corresponding decrease in serum cholesterol levels. 148 Additional suggested biomarkers include high sensitivity C-reactive protein, estrone sulfate, 149 and proteins involved in apoptotic pathways. 150 These types of clinical trials are invaluable for improving our ability to predict how statins may alter the progression of breast cancer, determine the extent to which individual breast cancer patients may respond to the statin administration, and identify appropriate biomarkers for examining their effectiveness. Whether or not these findings are applicable to other cancer types remains to be seen.
Should statins be used to prevent strokes
Strokes occur when blood flow to the brain is restricted, resulting in neuronal cell death. 151 This cell death often leads to the symptomatic dysfunction of parts of the body controlled by the affected brain regions. Strokes occur in the following two main forms: ischemic (more common) and hemorrhagic. The former is caused by a deficiency in blood flow characterized by blockage, whereas the latter is caused by damaging pressure produced by the bleeding of arterial blood vessels in the brain. Ischemic stroke can also be further subdivided into the following two causal categories: embolic stroke and thrombotic stroke. Thrombotic stroke is often associated with hypercholesterolemia and atherosclerosis and is caused by the formation of a blood clot in an artery that supplies the brain. 152
While statins have been considered for use in preventing the onset and severity of stroke, an analysis of the literature has produced weak and often inconsistent associations between stroke and hypercholesterolemia. 153 The inherent heterogeneity in the disease may be the likely culprit of these inconsistencies, as stroke has various etiologies. 154 However, it has been shown that statin-induced reduction in cholesterol concentrations decreases the risk of stroke in high-risk patient populations (ie, those with established coronary artery disease, hypertension, diabetes, or a high vascular risk).154,155 A meta-analysis 155 revealed that each 1 mM decrease in LDL cholesterol compared to a 21.1% overall reduction in the relative risk for stroke. These results were mirrored by an additional study showing a consistent 17%–21% reduction in incident stroke risk when patients were put on a statin regimen. 154
A multitude of recent experimental studies156–158 have pointed to a decrease in mortality risk with statin therapy when rendered both before and after the onset of stroke. 159 This decline in mortality risk may be attributed to the neuroprotective and neurorestorative effects of statins during the onset of an ischemic stroke and after, respectively.160–164 Furthermore, studies have shown that statin treatment has led to improvement in short-term and long-term poststroke quality of life and survival–-these results may even be modulated by the stroke severity.153,156,160–164 However, due to the inconclusive and discordant nature of some reports, more research must be conducted to validate the efficacy of statin therapy on stroke prevention and recovery.
Should statins be used to treat neurodegenerative disorders?
The use of statins to treat a number of neurodegenerative disorders, including Parkinson's and Alzheimer's diseases, has also been reported.165,166 The pathophysiology of Parkinson's disease involves aggregation of alpha-synuclein (α-S), a presynaptic, neuronal protein, which normally binds to plasma membranes. 167 In addition, high cholesterol levels are implicated in disease progression and the use of statins in animal models of Parkinson's disease can reduce α-S aggregation and neuropathology. 168 While some epidemiological studies report that statin use may reduce the risk of Parkinson's disease, 169 others imply that these protective effects are not observed after adjusting the results for cholesterol levels. 170
With respect to Alzheimer's disease, high cholesterol levels are associated with disease progression and the appearance of plaques, a key characteristic of Alzheimer's disease. 171 Despite promising results performed in animal models, the results from two randomized clinical trials failed to demonstrate a difference in cognitive decline in patients who were receiving statin treatment (simvastatin and/or pravastatin) for at least 12 months as compared to the placebo control. 172 Due to inconclusive evidence, statins and their role in the prevention and treatment of neurodegenerative diseases require further investigation.
Should statins be used to treat bacterial infections?
The ability of statins to modulate the immune and inflammatory responses may abrogate the pathogenesis of diseases, such as tuberculosis, sepsis, listeriosis, and infections of the host by a bacterial entity.173–177 With respect to bacterial infections, statins appear to modulate the expression of extracellular receptors involved in the attachment and uptake of certain bacteria. As shown in studies using Pseudonomas aerginousa, statins can impede bacterial motility and biofilm production as well as attenuate the secretion of proinflammatory cytokines. 176 Additional studies have been performed on several bacterial species, which have yielded consistent results.173,175 For example, a study involving Mycobacterium tuberculosis, the causative agent of most cases of tuberculosis, found that statins counteracted the bacteria's ability to inhibit immune cell maturation. 173 Similarly, when statins were administered prior to infection with Listeria monocytogenes, the ability of the bacteria to escape phagosomes was inhibited, which diminished their evasion of the immune response. 175 Although the precise mechanisms by which statins modulate the immune system in response to bacterial infections are unknown, these studies highlight the potential for the use of statins in the prevention and treatment of bacterial infections.
Should statins be used to treat patients with HIV-1?
Antiviral therapy is used extensively to treat individuals with HIV-1. 178 While pharmacologic agents, such as HIV-1 protease and nucleoside reverse transcriptase inhibitors, are effective at suppressing viral replication, their use is often accompanied by abnormal fat redistribution, peripheral fat wasting, central adiposity, and elevated cholesterol and triglyceride concentrations. 179 Thus, statins are often administered to HIV patients to reduce their cholesterol levels and normalize their body composition. 180 , 181 Statins may also reduce HIV-1 infection, presumably via inhibition of protein prenylation, downregulation of Rho activity, and rearrangement of the actin cytoskeleton, which prevents HIV-1 from gaining entrance to the host cells. 182 , 183 In a recent clinical study, statin administration to patients chronically infected with HIV-1 significantly reduced serum viral RNA loads. However, upon discontinuation of statin therapy, viral load rebounded. These data imply that future clinical studies should be conducted to further assess the use of statins as antiviral agents.
Conclusions
To fully understand whether statins display desirable pleiotropic effects and/or should be used as novel or adjunct therapies to treat various diseases–-aside from hypercholesterolemia–-a number of steps should be considered. First, the patients who are most likely to positively respond to the desired effect of statins must be identified. Second, the most effective dose, duration of use, and statin drug entity to be used must be clinically established. Third, biomarkers accurately reflecting the pleiotropic actions that are clearly indicative of a patient's response to the statin and are specific for the disease state of interest should be identified. Successful completion of these endeavors would then culminate in randomized clinical trials designed to evaluate the purported efficacy of various off-label effects of statins.
Abbreviations
alpha-synuclein;
ATP-binding cassette sub-family G member 2;
American College of Cardiology;
adverse effect;
American Heart Association;
AMP-activated protein kinase;
activator protein 1;
adenomatous polyposis coli;
atherosclerotic cardiovascular disease;
chronic obstructive pulmonary disease;
cardiovascular disease;
central nervous system;
cytochrome P4502C9/3A4;
estrogen receptor;
endothelial nitric oxide synthase;
extracellular signal-regulated kinase;
ETS protooncogene;
United States Food and Drug Administration;
guanosine triphosphate cyclohydrolase;
histone deacetylase;
human epidermal growth factor 2;
human immunodeficiency virus;
high-density lipoprotein;
3-hydroxy-3-methylglutaryl-coenzyme A;
3-hydroxy-3-methylglutaryl-CoA reductase;
inter cellular adhesion molecule 1;
inflammatory bowel diseases;
intermediate-density lipoproteins;
interferon β/γ;
janus kinase;
Kruppel-like factor 2;
low density lipoprotein cholesterol;
low density lipoprotein receptor;
lymphocyte function-associated antigen 1;
mitogen-activated protein kinases;
multiple sclerosis;
nuclear factor-kappa B;
nitric oxide;
odds ratio;
proprotein convertase subtilisin/kexin type 9;
phosphatidylinositol 3-inositol;
protein kinase A;
progesterone receptor;
phosphatase and tensin homolog;
rheumatoid arthritis;
Rho-associated protein kinase;
relapsing remitting multiple sclerosis;
statin-associated muscle symptoms;
solute carrier organic anion transporter family members 1B1/2B1;
systemic lupus erythematosus;
sterol regulatory element;
sterol regulatory element-binding protein;
signal transducer and activator of transcription 1/3/4;
type 2 diabetes mellitus;
type 1/2/17 helper cells;
regulatory T cells;
tumor suppressor p53;
very low-density lipoprotein.
Author Contributions
Wrote the first draft of the article: JTD, SFD, CEF, MFJ, VLN, JTO, APS, and HIS. Contributed to the writing of the article: JTD, SFD, CEF, MFJ, VLN, JTO, APS, and HIS. Agreed with the article results and conclusions: JTD, SFD, CEF, MFJ, VLN, JTO, APS, and HIS. Jointly developed the structure and arguments for the article: JTD, SFD, CEF, MFJ, VLN, JTO, APS, and HIS. Made critical revisions and approved the final version: JTD, SFD, CEF, MFJ, VLN, JTO, APS, and HIS. All authors reviewed and approved the final article.
Footnotes
Acknowledgments
We thank Ms Melanie Hurst, Mr Seth Kruger, and Ms Angeline Parente for their contributions during the initial phases of this work. We also thank Dr Robert Hadley and Dr Michael Piascik for their critical comments and helpful suggestions.