
Abstract
The proper localization and synthesis of postsynaptic glutamate receptors are essential for synaptic plasticity. Synaptic translation initiation is thought to occur via the target of rapamycin (TOR) and mitogen-activated protein kinase signal-integrating kinase (Mnk) signaling pathways, which is downstream of extracellular-regulated kinase (ERK). We used the model glutamatergic synapse, the Drosophila neuromuscular junction, to better understand the roles of the Mnk and TOR signaling pathways in synapse development. These synapses contain non-NMDA receptors that are most similar to AMPA receptors. Our data show that Lk6, the Drosophila homolog of Mnk1 and Mnk2, is required in either presynaptic neurons or postsynaptic muscle for the proper localization of the GluRIIA glutamate receptor subunit. Lk6 may signal through eukaryotic initiation factor (eIF) 4E to regulate the synaptic levels of GluRIIA as either interfering with eIF4E binding to eIF4G or expression of a nonphosphorylatable isoform of eIF4E resulted in a significant reduction in GluRIIA at the synapse. We also find that Lk6 and TOR may independently regulate synaptic levels of GluRIIA.
Introduction
Glutamate mediates the majority of neurotransmission in the central nervous system (CNS). 1 Once a synapse is established, its strength is modulated by changes in presynaptic glutamate release, synaptic size, and number of postsynaptic receptors.2,3 The capacity of the synapse to change as a result of synaptic activity, or synaptic plasticity, requires the translation of proteins that modulate synapse function. 4 7 Although synaptic plasticity is initially mediated by changes in protein trafficking, translation of synaptic mRNAs is required for structural changes to the synapse and to maintain the altered neurotransmission. 8 The importance of local translation for synaptic plasticity is illustrated by aberrant translation initiation, which is thought to occur in fragile X syndrome as a result of the loss of function of the translational repressor, fragile X mental retardation protein. 9
Protein synthesis is primarily regulated at the step of translation initiation, 10 which requires the binding of eukaryotic initiation factor (eIF) 4E to the 5′7-methylguanosine cap (5′ cap) of mRNA.11,12 This binding recruits eIF4G to eIF4E in a process that facilitates ribosome binding. eIF4G provides a scaffold for the binding of eIF4A to form the eIF4F complex. 13 Two growth factor-activated pathways, including the mitogen-activated protein kinase (MAPK) and mammalian target of rapamycin (mTOR) pathways, regulate translation initiation by influencing the capacity of eIF4E to bind to the 5′ cap.
MAPKs are involved in a variety of cellular processes, including, but not limited to, cellular proliferation, apoptosis, differentiation, transformation, and cell movement.14,15 MAPK signaling can occur via three divergent pathways, including extracellular-regulated kinase (ERK), p38, and c-Jun N-terminal kinase signaling. 16 Each of the MAPKs is a Ser/Thr kinase activated by phosphorylation via upstream MAPK kinases.14,15 ERK is activated by synaptic depolarization, calcium influx, and neurotrophin signaling.17,18 MAPK signal-integrating kinases (Mnks), including Mnk1 and Mnk2, are subsequently phosphorylated and activated by ERK.19,20 Once phosphorylated, Mnks regulate translation initiation by phosphorylating eIF4E to influence translation initiation. 21 23
Similar to the Mnks, mTOR is a highly conserved Ser/Thr kinase involved in several cellular processes, such as cellular homeostasis and proliferation. mTOR forms two different complexes, one of which, mTORC1, phosphorylates S6 kinase 1 to promote the synthesis of ribosomal proteins 24 and phosphorylates 4E-binding protein (4E-BP) to inhibit its binding to eIF4E. 25 Thus, the mTOR pathway represses the actions of a translational inhibitor, 4E-BP, thereby promoting the interaction between eIF4E and eIF4G. 26
Both of these pathways regulate synaptic plasticity by influencing translation initiation. Mnk activation downstream of brain-derived neurotrophic factor (BDNF) signaling enhances long-term potentiation (LTP) using distinct mechanisms at two different time points to enhance translation in dentate gyrus cells. 17 BDNF signaling through Mnk1 was subsequently shown to increase the levels of proteins required for vesicle trafficking and exocytosis in cortical neurons. 27 Rapamycin, an inhibitor of mTORC1, 28 suppresses BDNF-induced late LTP in rat hippocampal neurons. 29 These data indicate that both mTOR and Mnk signaling enhance synaptic plasticity, but the mechanisms utilized by these pathways are unclear.
We used the Drosophila neuromuscular junction (NMJ) to investigate the mechanisms by which Mnk and TOR regulate the synaptic levels of the glutamate receptor (GluR) subunit, GluRIIA. Drosophila expresses a single Mnk homolog, Lk6, which phosphorylates eIF4E 30 and regulates growth. 31 The Drosophila NMJ is a glutamatergic synapse, which is structurally and functionally similar to mammalian CNS glutamatergic synapses, and contains AMPA-like, non-NMDA receptors. 32 One NMJ postsynaptic GluR subunit, GluRIIA, is locally translated in an eIF4E-dependent manner as a result of synaptic activity. 33 We show, for the first time, that Lk6 regulates glutamatergic neurotransmission and synaptic localization of GluRIIA. Lk6 is required in either presynaptic neurons or postsynaptic muscles for proper GluRIIA localization. Inhibiting the interaction between eIF4E and eIF4G pharmacologically or overexpressing a nonphosphorylatable isoform of eIF4E in these tissues also resulted in the loss of GluRIIA from the NMJ indicating that Lk6 may signal through eIF4E to regulate GluRIIA localization. Lk6 may regulate the synaptic levels of GluRIIA independent of the TOR complex as simultaneous inhibition of Lk6 and TOR signaling resulted in an additive loss of synaptic GluRIIA.
Materials and Methods
Fly stocks
Fly stocks were maintained at 25°C in vials containing Jazz-Mix Drosophila food (Fisher Scientific). The UAS-GAL4 system was used for tissue-specific expression. 34 All fly stocks were obtained from the Bloomington Drosophila Stock Center except lk6 RNAi , which was obtained from the Vienna Drosophila RNAi Center, and Dcr2;;elav-Gal4 and Dcr2;;24B-Gal4, which were gifts from Aaron DiAntonio's Lab. w 1118 and outcrossed controls were used as controls for each experiment as appropriate.
Immunohistochemistry and confocal microscopy
Third-instar larvae were dissected on Sylgard plates in Roger's Ringer solution (135 mM NaCl, 5 mM KCl, 4 mM MgCl2-6H2O, 1.8 mM CaCl22H2O, 5 mM TES, and 72 mM sucrose) supplemented with 2 mM glutamate. 35 Larvae were fixed for 30–45 minutes in either Bouin's fixative (for GluR and Bruchpilot [Brp] antibodies) or 4% paraformaldehyde in phosphate buffered saline (PBS) (for all other antibodies). After fixation, larval dissections were washed in PTX (PBS + 0.1% Triton) and PBTX (PBS + 0.1% Triton + 1% bovine serum albumin [BSA]). Primary antibodies were applied overnight at 4°C in PBTX. Mouse α-Brp (aka nc82, 1:50), mouse α-discs large (DLG) (1:1000), mouse α-synaptotagmin (Syt) (1:100), and mouse α-GluRIIA (1:100) were acquired from the Developmental Studies Hybridoma Bank. Rabbit α-GluRIIB (1:2000) and rabbit α-GluRIIC (1:5000) were generous gifts from Aaron DiAntonio. 36 Mouse α-acetylated tubulin (1:1000) and phalloidin (1:200) were obtained from Sigma-Aldrich and Invitrogen, respectively. After washing the larval preparations with PBTX, additional antibodies including horseradish peroxidase (HRP) (1:125, Jackson ImmunoResearch) and species-specific fluorescein isothiocyanate (FITC) (1:400, Jackson ImmunoResearch) were applied for two hours at room temperature. Larvae were washed with PBTX before mounting on slides with Vectashield (Vector Laboratories).
Images of third-instar larval 6/7 NMJs from left or right hemisegments at A3 or A4 were acquired using the 60× objective of an Olympus FV1000 confocal microscope. Imaging parameters were established for controls and subsequently used for all experimental animals. Approximately equal numbers of control and experimental animals were imaged during each imaging session.
Electrophysiology
Third-instar larvae were filet dissected at room temperature on Sylgard-coated coverslips and glued down with Vetbond Tissue Adhesive (World Precision Instruments). Dissections and recordings were performed in Roger's Ringer solution. Muscle 6 of hemisegments 3 or 4 was clamped at –60 mV using an Axoclamp 900A amplifier (Molecular Devices). Current injecting and recording electrodes were filled with 3 M KCl and were used provided their resistances were 10–20 MΩ. Segmental nerves were stimulated with an electrode filled with bath saline. A 0.5 Hz, 10 V stimulus was delivered using a Grass S88 stimulator with a SIU5 isolation unit (Grass Technologies). Recordings were digitized using a Digidata 1443 digitizer (Molecular Devices). pClamp software (version 10.4) was used for data analyses. Quantal content was calculated by dividing the eEJC area (nA × ms) by the mEJC area (nA × ms) for each animal. An equal number of control and experimental recordings were acquired each day.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
RNA was extracted from 8 to 12 third-instar larvae using TRIzol (Invitrogen) as previously described. 37 Single-plex reactions using gene-specific primers for gluRIIA, gluRIIB, gluRIIC, or GAPDH and the iTaq Universal SYBR Green One-Step Kit (Bio-Rad) were performed using a Stratagene Mx3000P qPCR System (Agilent Technologies). 100 ng of total RNA was added to each reaction. Three technical replicates and two biological replicates were performed for each reaction. ΔC(t) values were obtained by subtracting the GAPDH C(t) value from the gluR C(t) value.
4EGI-1 and rapamycin treatments
Control and lk6 mutant larvae were reared for four days in vials as previously described. After four days, wandering third-instar larvae were placed on nutrient-rich apple juice agar plates containing yeast supplemented with 10 μM 4EGI-1 (Fisher Scientific) or yeast containing an equal volume of dimethyl sulfoxide (DMSO). Larvae remained on plates for 24 hours and then were used for dissection and immunohistochemistry. 10 μM 4EGI-1 was previously shown to inhibit cap-dependent translation when fed to flies. 38 We also found that 10 μM 4EGI-1 did not significantly affect larval viability (Supplementary Fig. 1).

Lk6 promotes the synaptic localization of GluRIIA. (
Parental genotypes, including w 1118 and lk6 RNAi , and flies containing the tissue-specific drivers Actin5c-Gal4 (for expression in all tissues), Dcr2;;elav-Gal4 (for expression in neurons), and Dcr2;;24B-Gal4 (for expression in postsynaptic muscles), were placed on apple juice agar plates 39 with a yeast mix for 24 hours. After 24 hours, the parental generation was removed and F1 offspring remained on the plates. After 96 hours, the yeast mix was replaced with a yeast mix containing either 0.1 mM rapamycin (Fisher Scientific) dissolved in ethanol or yeast mix containing an equal volume of ethanol for an additional 24 hours. This concentration of rapamycin was selected based on the previous experiments40,41 and to standardize the size and optimize the viability of the animals. Third-instar larvae were used 24 hours after changing the yeast mix for dissection and immunohistochemistry.
Image analyses and statistics
Compressed images of confocal micrograph z-series were used for data analyses. GluRIIA cluster sizes were obtained by measuring the area of GluRIIA puncta 42 localized immediately adjacent to or in direct opposition of the presynaptic motor neuron (as determined by HRP immunolabeling) using National Institutes of Health's ImageJ. Brp densities were calculated by counting the number of Brp puncta and dividing by the area of the presynaptic motor neuron. Relative fluorescence intensities were quantified by manually tracing around the NMJ, recording the mean fluorescence for the area and fluorophore-specific channel using the Adobe Photoshop (CS6 version 13), and then subtracting the background from a nonsynaptic area of equal size. For tubulin and phalloidin quantifications, a nonsynaptic and nonmuscle area was used to determine the background. Morphology of the presynaptic motor neuron was determined by manually counting the boutons and branches. Branches were defined as bifurcations of the motor neuron containing at least two boutons.
Statistical analyses of the data were performed using GraphPad Prism (version 5.00). Student's t-tests were used to analyze two data sets. One-way analysis of variance using Tukey's (equal variance between data sets) or Holm-Sidak's (significant difference in variance between data sets) post hoc tests were used to analyze the potential differences between more than two data sets. In all figures, the level of statistical significance is as follows: ∗P ≤ 0.05, ∗∗P ≤ 0.001, and ∗∗∗P ≤ 0.0001. Error bars represent the standard error of the mean. Summary statistics for all data are reported in Supplementary Table 1.
Results
Lk6 promotes the synaptic localization of postsynaptic GluRs and synaptic transmission
Translation initiation is required for several forms of synaptic plasticity, including LTP,4,5 long-term depression,43,44 and memory consolidation.6,7 We found, through a forward genetic screen, that the Drosophila homolog of Mnk1 and Mnk2, Lk6, regulates the localization of GluRs to the NMJ. These glutamatergic synapses are similar to mammalian central glutamatergic synapses both at the subcellular and molecular levels. 45 Drosophila NMJ GluRs, similar to AMPA receptors, are tetramers 46 that mediate fast synaptic transmission. 47 Lk6 is 57% identical and 74% similar to Mnk1 (using NP_001272416.1) and 58% identical and 73% similar to Mnk2 (using NP_067437.2) with slightly higher sequence conservation in the Lk6 catalytic domains. 48 Mnks are activated by MAPKs 20 and regulate translation initiation by phosphorylating eIF4E21–23,49 thereby influencing assembly of the eIF4F cap-binding complex.
In order to better understand the role of Lk6 in glutamatergic synapse development, we characterized the lk6 mutant phenotype using the lk6 2 hypomorph. The lk6 2 mutant contains a deletion of all exons downstream of the first intron caused by the excision of an EP P-element producing a loss of function allele. 30 Drosophila NMJ GluRs contain either the GluRIIA or GluRIIB subunits along with three essential subunits, including GluRIIC, GluRIID, and GluRIIE.36,46 Both GluRIIA 50 and GluRIIB 51 are localized exclusively to postsynaptic muscle cells. Lk6 2 mutants exhibited a significant decrease in GluRIIA cluster size (Fig. 1A and B) but no significant differences in GluRIIB (Fig. 1D) or GluRIIC (Fig. 1E) cluster sizes compared to ω1118 controls. Cluster size measurements were used because they correlate with the function of the synapse. 42 We confirmed the loss of synaptic GluRIIA in lk6 36 mutants (GluRIIA cluster sizes, w1118: 1.14 ± 0.06 μm2, n = 120 clusters from 12 animals; lk6 36 : 0.70 ± 0.05 μm2, n = 140 clusters from 14 animals; P < 0.0001), which contain a His to Arg substitution at amino acid 154. 31 Mutations in lk6 did not affect NMJ morphology (Fig. 1C).
Mnks may indirectly affect transcription by regulating mRNA localization of transcriptional regulators. 52 To determine whether mutations in lk6 impact the synaptic localization of GluRs by affecting transcription of gluR subunits, we measured gluR subunit mRNA levels using qRT-PCR. Interestingly, gluRIIA and gluRIIB mRNA levels were significantly increased in lk6 2 mutants compared to controls, while there were no significant differences in gluRIIC mRNA levels (Fig. 1F).
The microtubule 53 55 and actin cytoskeletons 56 58 enable GluR trafficking and synaptic localization. Furthermore, Mnk1 59 and the 220 kDa isoform of Lk6 60 are colocalized with centrosomes, structures composed of microtubules. Therefore, we investigated the possibility that Lk6 may affect GluR trafficking to the synapse by affecting cytoskeletal dynamics. We examined the microtubule cytoskeleton by immunolabeling acetylated tubulin, which demarcates stable microtubules. 61 There were no significant differences in the levels of synaptic or muscle acetylated tubulin in lk6 2 mutants compared to controls (Supplementary Fig. 2A). Similarly, we did not detect any gross morphological differences in the sarcomeric structure of lk6 2 mutant muscles as indicated by labeling F-actin with phalloidin. 62 We also did not observe alterations in axon targeting, muscle patterning, or muscle sizes in lk6 2 mutants compared to controls (Supplementary Fig. 2B).

Lk6 positively regulates synaptic transmission. Spontaneous miniature (mEJCs) and evoked endplate junctional currents (eEJCs) were recorded from muscle 6 of third-instar larvae after the muscle was clamped at –60 mV. (
Reductions in the number of postsynaptic GluRs may occur as a result of changes in presynaptic glutamate release 42 or extracellular glutamate concentrations. 63 To assess the possibility that Lk6 may affect synaptic glutamate availability thereby altering the localization of postsynaptic GluRs, we used two complimentary approaches. First, we used immunocytochemistry to examine synaptic proteins that influence neurotransmitter release and organization of postsynaptic protein complexes. There were no significant differences in the synaptic levels of DLG, a postsynaptic scaffolding protein that regulates synaptic development, 64 or the presynaptic proteins Brp and Syt, which are localized to active zones65,66 and regulate presynaptic neurotransmitter release, 67 respectively (Supplementary Fig. 3), in lk6 2 mutants compared to controls.
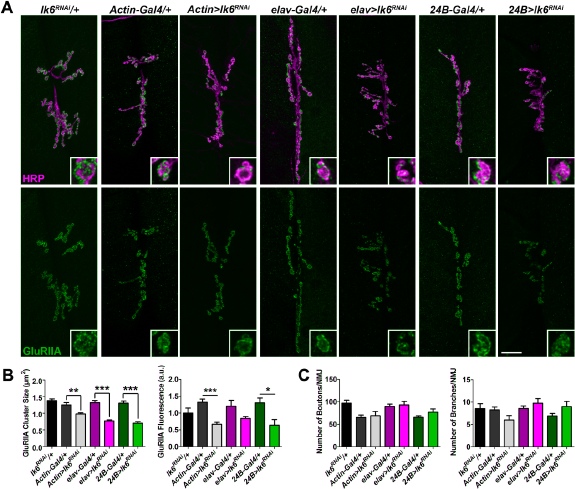
Lk6 is important both pre- and postsynaptically for GluRIIA localization. (
Next, we used two-electrode voltage clamp to further investigate whether Lk6 regulates presynaptic glutamate release. Evoked endplate junction current (eEJC) amplitudes were significantly reduced in lk6 2 mutants (Fig. 2A and B) compared to w1118 controls. Similarly, spontaneous neurotransmission, as measured by miniature EJC (mEJC) amplitudes, were also significantly reduced in lk6 2 mutants (Fig. 2C–E). There were no significant differences, however, in quantal content or mEJC frequency (Fig. 2B and D). Interestingly, there was a significant reduction in mEJC rise time in lk6 2 mutants suggesting that Lk6 regulates GluR channel kinetics likely by influencing the synaptic localization of the GluRIIA subunit. These data indicate that Lk6 may primarily affect postsynaptic GluRs without affecting presynaptic glutamate release. Collectively, our data are consistent with a loss of synaptic GluRs in lk6 mutant synapses resulting in an attenuated response to evoked stimuli and spontaneous neurotransmission.
Lk6 regulates GluRIIA localization by both pre- and postsynaptic mechanisms
Our efforts to detect Lk6 at the NMJ using an antibody 60 were unsuccessful. Therefore, we assessed the tissue-specific contributions of Lk6 to GluR localization using a combination of knock down and rescue experiments. We knocked down lk6 by expressing an UAS-lk6 RNAi transgene under the control of the Actin5c-Gal4 (all tissues), Dcr2;;elav-Gal4 (neurons), or Dcr2;;24B-Gal4 (muscle) drivers. Knock down of lk6 in all tissues, neurons, or muscles produced a significant reduction in GluRIIA cluster sizes compared to the outcrossed controls as determined by a one-way analysis of variance (Supplementary Table 1). There was also a significant reduction in relative GluRIIA fluorescence intensity when lk6 was knocked down in all tissues or in postsynaptic muscles compared to the outcrossed controls. Although there was a trend toward reduced GluRIIA fluorescence when lk6 was knocked down in neurons, it was not significant (Fig. 3A and B). This may indicate that, while the individual GluRIIA clusters are smaller, there may be a slight increase in the number of clusters. Knock down of lk6 in any tissue type did not alter the morphology of presynaptic motor neurons (Fig. 3C). These data indicate that Lk6 is important in both presynaptic neurons and postsynaptic muscle for the proper localization of GluRIIA-containing receptors.
We next performed rescue experiments to determine whether Lk6 is required in pre- or postsynaptic cells or both cell types for GluR localization. An UAS-lk6 transgene was expressed in the lk6 2 mutant background specifically in neurons (using the elav-Gal4 driver) or muscles (using the 24B-Gal4 driver). Expression of lk6 in either neurons or muscles rescued GluRIIA cluster sizes and GluRIIA fluorescence to near w1118 control levels (Fig. 4A and B). These data show that the loss of GluRIIA in lk6 2 mutants can be attributed specifically to the loss of Lk6. Consistent with our knock down data, our rescue experiments indicate that Lk6 is required in presynaptic motor neurons or postsynaptic muscles for the synaptic localization of GluRIIA.

Lk6 is required in either presynaptic neurons or postsynaptic muscle for proper GluRIIA localization. (
Lk6 may signal through eIF4E to regulate synaptic levels of GluRIIA
Lk6 is activated by ERK signaling but not by p38 MAPK signaling. Lk6 phosphorylates eIF4E at Ser251, 48 which is analogous to Ser209 in mammals, the residue phosphorylated by Mnk1. 20 Mnk-mediated phosphorylation of eIF4E reduces its interaction with the 5′ cap,68,69 but this may enhance translation by liberating eIF4E from eIF4G to enable additional rounds of translation initiation. 70 Our data demonstrate that Lk6, localized in both neurons and muscles, is important for synaptic levels of GluRIIA (Figs. 3 and 4). Given the role of Lk6 in translation initiation, we hypothesized that Lk6 positively regulates translation initiation of the gluRIIa subunit transcript, which has been shown to be locally translated at the NMJ, 33 by phosphorylating eIF4E. To test this hypothesis, we first disrupted the interaction between eIF4E and eIF4G and then expressed a nonphosphorylatable isoform of eIF4E encoded by the UAS-eIF4ES251A construct 31 to determine if these manipulations would alter the localization of GluRIIA to the synapse.
Formation of the eIF4F complex is regulated by 4E-BP, which binds to eIF4E preventing the interaction of eIF4E with eIF4G. 25 Similarly, 4EGI-1 is a reversible competitive inhibitor of eIF4E. 4EGI-1 specifically binds to and blocks the eIF4G motif of eIF4E without affecting the binding of 4E-BP. 71 Interfering with eIF4F complex formation by feeding larvae 10 μM 4EGI-1 resulted in a significant reduction in GluRIIA cluster sizes and relative GluRIIA fluorescence intensity in w1118 controls but not in lk6 2 mutants (Fig. 5A and B). Inhibition of the eIF4E-eIF4G interaction, however, did not significantly affect NMJ morphology (Fig. 5C). The loss of GluRIIA in w1118 but not in lk6 2 mutants after inhibiting the binding of eIF4G to eIF4E indicates that Lk6 may promote the functional interaction of eIF4E and eIF4G to positively regulate the synaptic localization of GluRIIA.
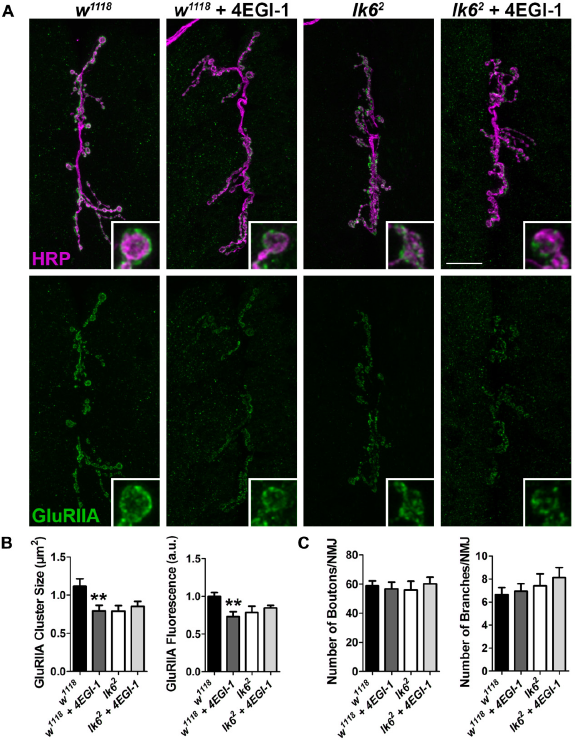
Disrupting the interaction between eIF4E and eIF4G negatively affects synaptic GluRIIA in w1118 controls but not in lk6
2
mutants. 10 μM 4EGI-1 was fed to larvae to interfere with eIF4G binding to eIF4E. (
Similarly, expression of eIF4ES251A ubiquitously, presynaptically, or postsynaptically resulted in significantly decreased GluRIIA cluster sizes compared to outcrossed controls (Fig. 6A and B). The expression of eIF4ES251A in neurons or muscle resulted in a significant reduction in GluRIIA relative fluorescence intensity (Fig. 6A and B). Although there was a trend toward reduced GluRIIA fluorescence, when the UAS-eIF4ES251A transgene was expressed in all tissues, it was not significant (Supplementary Table 1). When eIF4ES251A was expressed ubiquitously, there were no significant differences observed in the morphology of the presynaptic motor neuron (Fig. 6C). The expression of eIF4ES251A either presynaptically or postsynaptically, however, differentially affected the morphology of the presynaptic motor neuron. When eIF4ES251A was expressed in presynaptic neurons but not in postsynaptic muscles, there was a significant increase in bouton number. Conversely, the expression of eIF4ES251A in muscles but not in presynaptic neurons resulted in a significant reduction in the number of NMJ branches (Fig. 6C). Taken together, our data suggest that Lk6 may phosphorylate eIF4E to promote the synaptic localization of GluRIIA and indicate that translation efficiency is important for presynaptic motor neuron morphology.

Expression of a nonphosphorylatable eIF4E impairs the synaptic localization of GluRIIA. An UAS-eIF4ES251A transgene was expressed in all tissues using the Actin5c-Gal4 driver, in neurons using the elav-Gal4 driver, or in postsynaptic muscles using the 24B-Gal4 driver. (
Lk6 and TOR may independently regulate the synaptic localization of GluRIIA
Translation initiation in Drosophila is regulated by both Lk6 and TOR, which phosphorylates 4E-BP thereby increasing the availability of eIF4E for translation initiation. 26 In order to establish a molecular model for GluR localization, we sought to test whether we would observe a similar impact on synaptic GluR localization by inhibiting TOR activity at its FKBP-rapamycin binding domain using rapamycin. 72 Because Lk6 and TOR use different mechanisms to influence eIF4E availability, we hypothesized that either inhibition of Lk6 or TOR function would affect the localization of synaptic GluRs. To test this, we again knocked down lk6 by expressing UAS-lk6 RNAi in all tissues using the Actin5c-Gal4 driver, in presynaptic neurons using the Dcr2;;elav-Gal4 driver, or in postsynaptic muscle using the Dcr2;;24B-Gal4 driver and simultaneously inhibited TOR signaling using rapamycin. Knock down of lk6 in all tissues, presynaptic neurons, or post-synaptic muscles coupled with TOR inhibition resulted in a significant decrease in mean GluRIIA cluster sizes and relative GluRIIA fluorescence compared to knock down only animals (Fig. 7A and B). TOR inhibition coupled with the knock down of lk6 in all conditions examined resulted in a significant overgrowth of the presynaptic motor neuron (Fig. 7C). Thus, TOR signaling but not Lk6 signaling restrains growth of the NMJ. These data suggest that Lk6 and TOR signaling independently regulate the synaptic localization of GluRIIA, possibly by each regulating different intermolecular interactions required for translation initiation.
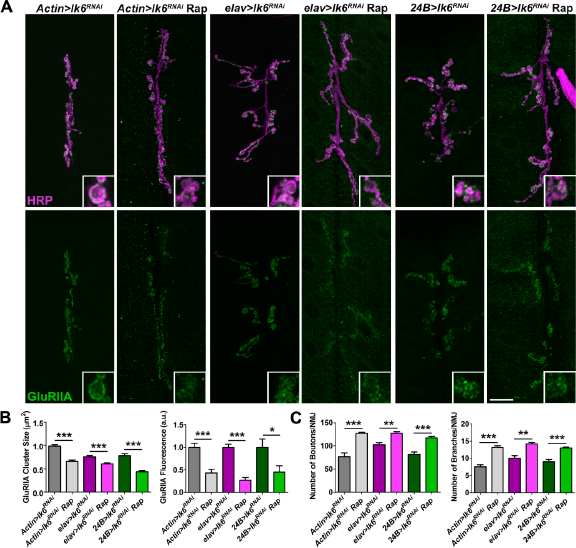
Knock down of lk6 coupled with the inhibition of TOR signaling results in a greater loss of synaptic GluRIIA. Lk6 was knocked down in all tissues using the Actin5c-Gal4 driver, in neurons using the Dcr2;;elav-Gal4 driver, or in postsynaptic muscles using the Dcr2;;24B-Gal4 driver. TOR signaling was inhibited by feeding animals 100 μM of rapamycin in a yeast mix for 24 hours. (
Discussion
Translation of synaptic mRNAs is required for synaptic plasticity,4–7,43,44 but the role of important translation initiators including the Mnks in synaptic development is largely unexplored. We found that the Drosophila homolog of Mnk1 and Mnk2, Lk6, 30 is required in either pre- or postsynaptic cells for the synaptic localization of GluRs. Lk6 may regulate gluRIIa translation as the loss of synaptic GluRIIA also occurs as a result of expression of an isoform of eIF4E that cannot be phosphorylated by Lk6 but does not occur when the interaction between eIF4E and eIF4G is disrupted in lk6 2 mutants. Lk6 likely works in parallel with TOR signaling to regulate the synaptic localization of GluRIIA (Fig. 8). Our collective data demonstrate that Lk6 is essential for synapse development and function.

Model for the translation of synaptic mRNAs. Lk6 and TOR signaling converge on eIF4E to regulate translation initiation in postsynaptic cells. These pathways differentially promote GluRIIA expression and/or localization.
Lk6 is required for the synaptic localization of GluRIIA
Postsynaptic GluRs cluster in apposition to presynaptic release sites. 51 The GluRIIA subunit increases the open time of the receptor leading to larger mEJPs 47 and is locally translated as a result of synaptic activity. 33 Synaptic levels of GluRIIA but not GluRIIB or GluRIIC are significantly reduced in lk6 2 mutants (Fig. 1). The loss of GluRIIA impairs synaptic function as the amplitudes of both evoked and spontaneous neurotransmission are attenuated in lk6 2 mutants (Fig. 2). Our data do not support a dominant pre- or postsynaptic role for Lk6 at the synapse. The reduction in mEJC amplitudes could be attributed to the postsynaptic loss of GluRIIA-containing receptors at the synapse of lk6 2 mutants, but it could also be attributed to a reduction in the size of presynaptic vesicles. 73 Although we did not observe a significant difference in quantal content, an indicator of the number of presynaptic vesicles released, 51 in lk6 2 mutants, this may be due to a compensatory increase in the number of vesicles released as a result of the selective loss of GluRIIA as occurs in GluRIIA mutants. 47
Our data instead suggest that Lk6 is required in either presynaptic neurons or postsynaptic muscle for synaptic GluR localization (Fig. 4). Knock down of lk6 in either pre- or postsynaptic cells resulted in the mislocalization and/or loss of synaptic GluRIIA (Fig. 3). Although we did not observe differences in the density of Brp or synaptic levels of Syt (Supplementary Fig. 3), other synaptic mechanisms influence postsynaptic GluR localization. Bidirectional synaptic signaling may be altered in lk6 2 mutants. For example, acute BDNF treatment increased the surface expression of the AMPA receptor subunits, GluA1 and GluA2, as a result of ERK signaling. 74 BDNF is released from both presynaptic neurons and postsynaptic dendrites and binds to tropomyosin-related kinase B receptors localized both pre- and postsynaptically. BDNF signaling enhances presynaptic glutamate release and levels of the postsynaptic proteins GluA1, GluA2, and GluA3, along with the scaffolding proteins SAP97, GRIP1, and PICK1. 75 Drosophila expresses two neurotrophins, DNT1 and DNT2, which are similar in sequence and structure to BDNF. 76 DNT1 and DNT2 are secreted from postsynaptic muscles, but the receptors for these neurotrophins have not yet been identified. 77 Therefore, a similar mechanism may exist at the Drosophila NMJ where bidirectional neurotrophin signaling positively affects both presynaptic glutamate release and the levels of postsynaptic GluRs and this mechanism may be attenuated in lk6 2 mutants.
An additional possibility is that Lk6 may regulate communication between pre- and postsynaptic cells by acting on cell adhesion molecules (CAMs). CAMs are transmembrane proteins that stabilize the connection between the presynaptic neuron, the postsynaptic cell, and/or the glial cell. In the mature synapse, most CAMs are centrally localized 78 and help organize the synaptic protein network of their respective cell. 79 CAM signaling is activated by binding to themselves, other CAMs, or the extracellular matrix and is important for synaptic plasticity and the localization of neurotransmitter receptors.80,81 Attractive potential targets of Lk6 include the neurexin-neuroligin complex, which, at mammalian CNS synapses, recruits GluA2 to the synapse through interactions with PICK1 82 and has been shown to regulate GluRIIA localization in Drosophila embryos. 83 Lk6 may also regulate translation of cadherin-catenin complexes, which, in mammals, have been shown to enhance the surface localization of GluA2 84 and the kainate receptor subunit, GluK6. 85 In support of this, Mnk1 has been shown to regulate the translation of laminins, which are extracellular matrix ligands of integrin receptors, and Neurexin-1. 27
Lk6 may regulate synaptic levels of GluRIIA by a non-canonical mechanism or by directly regulating translation of gluRIIa subunit mRNA
Lk6 is phosphorylated by ERK and phosphorylates eIF4E 48 at Ser251, which is analogous to Ser209 in mammals. 86 Exactly how this phosphorylation regulates translation is not well understood. While an early report found that phosphorylation of eIF4E enhanced its affinity for the 5′ cap, 87 other reports indicated that Mnk-induced phosphorylation of eIF4E reduced its interaction with the 5′ cap.68,69 Disrupting the interaction between eIF4E and the 5′ cap, however, may enhance translation efficiency by increasing the availability of eIF4E to enable additional rounds of translation initiation. 70 Similarly, Mnk-mediated phosphorylation of eIF4E has been shown to either enhance88,89 or repress90,91 translation initiation.
Our data indicate that the loss of synaptic GluRIIA in lk6 2 mutants may be translation dependent. Interfering with the association between eIF4E and eIF4G using the competitive inhibitor, 4EGI-1, produced a significant decrease in synaptic GluRIIA (Fig. 5) in controls but not in lk6 2 mutants. Similarly, expression of a nonphosphorylatable mutant isoform of eIF4E (eIF4ES251A) in either presynaptic neurons or postsynaptic muscle resulted in the loss of GluRIIA from the synapse (Fig. 6). Two hypotheses emerge from these data. First, Lk6 and eIF4E may independently regulate the synaptic localization of GluRIIA. The loss of GluRIIA is more pronounced after the expression of UAS-eIF4ES251A than UAS-lk6RNAi in both presynaptic neurons and postsynaptic muscle (compare the quantification in Figs. 3 and 6). We attempted to test this hypothesis by performing epistatic analyses to delineate the relationship between Lk6 and eIF4E. Consistent with previous reports, 31 however, overexpression of lk6 or expression of a constitutively active isoform of Lk6 phenocopies lk6 loss-of-function phenotypes (data not shown).
Lk6 could phosphorylate other substrates in addition to eIF4E. In support of this hypothesis, Mnk1 phosphorylates a variety of substrates, including polypyrimidine tract-binding protein-associated splicing factor (PSF) 92 in addition to eIF4E 20 and eIF4G. 21 Phosphorylation of PSF increases the association between PSF and AU-rich elements (AREs), 92 which are sequences in the 3′-untranslated region that regulate the stability of mRNAs as a result of interactions with AU-rich element-binding proteins. 93 In addition, Mnk1 phosphorylates phospholipase A2, enhancing its catalytic activity. 94 Phospholipase A2 regulates membrane fusion events 95 and promotes LTP96,97 and the plasma membrane localization of AMPA receptors. 98
Second, the loss of GluRIIA in lk6 2 mutants may be the result of deficient translation initiation of gluRIIa itself or synaptic proteins that properly localize GluRIIA. In support of this hypothesis, Mnks 23 and Lk6 31 are not global regulators of translation initiation. Synaptic GluRIIA levels are positively correlated with eIF4E levels in the Drosophila NMJ postsynaptic subsynaptic reticulum.33,99 Genheden et al (2015) recently identified 718 proteins whose synthesis was altered after BDNF treatment in Mnk knock out primary neurons compared to wild-type neurons. Gene ontology analysis indicated that proteins important for vesicle release and/or trafficking and synaptic plasticity are translated after BDNF treatment in a Mnk-dependent manner. 27
The Lk6/Mnk pathway may function in parallel to the TOR signaling pathway to regulate the synaptic localization of GluRIIA
TOR signaling is important for synaptic plasticity 100 and regulates translation initiation by phosphorylating 4E-BP leading to its dissociation from eIF4E. 26 Loss of 4E-BP2 function increases GluA1 and GluA2 levels but not NMDA receptor subunit levels and enhances AMPA receptor-dependent neurotransmission. 101 Thus, TOR signaling may work cooperatively with Lk6 to regulate translation. This model is supported by data from mammalian cells where mTOR phosphorylation of 4E-BP 26 increases the binding of eIF4E to eIF4G and the 5′ cap. 102 Mnks then interact with eIF4G to phosphorylate eIF4E. 21 Components of both the ERK-Mnk-eIF4E and phosphoinositol 3-kinase-Akt-mTOR pathways are localized to synapses with eIF4E enriched in the postsynaptic density. 103 Similarly, eIF4E is localized to the postsynapse at the Drosophila NMJ, 33 and Akt specifically regulates GluRIIA localization and mEJC amplitudes. 104
We observed a greater reduction in the synaptic localization of GluRIIA when TOR signaling was inhibited and lk6 was knocked down compared to animals with intact TOR signaling but knocked down lk6 (Fig. 6). These data could indicate that both TOR and Lk6 regulate translation, and this regulation is, at least partly, independent of the other pathway. In agreement with this, Mnk inhibitors in combination with rapamycin more severely attenuate protein synthesis compared to either Mnk inhibition or rapamycin alone. 88 Although we might expect to see a more severe reduction in synaptic GluRs after the inhibition of TOR signaling coupled with lk6 knock down, there are a few probable explanations for these results. First, a number of studies suggest that rapamycin treatment only partially inhibits 4E-BP phosphorylation. 105 107 Therefore, some level of translational initiation could proceed uninhibited. Second, the inhibition of mTORC1 signaling by rapamycin may activate Mnk2, which then phosphorylates eIF4E.108,109 Finally, neuronal and synaptic mRNAs have been shown to be translated via both cap-dependent and cap-independent mechanisms.110,111
Conclusion
Lk6 is required in presynaptic neurons or postsynaptic muscle cells for the proper synaptic localization of the GluRIIA subunit. Our collective results support a model whereby Lk6 regulates GluRIIA localization through molecular interactions with eIF4E (Fig. 8). Our data suggest that Lk6 and TOR signaling occur in parallel and may converge to regulate the eIF4E activity. Disruptions within these pathways have differential negative effects on GluR localization. These data further our understanding of the mechanisms that regulate GluR localization and provide insight on the contribution of translational regulators to synapse development.
Author Contributions
Conceived and designed the experiments: NAH and FLWL. Analyzed the data: NAH, TLD, and FLWL. Wrote the first draft of the article: FLWL. Contributed to the writing of the article: NAH, BLT, and FLWL. Agreed the article results and conclusions: NAH, TLD, BLT, and FLWL. Jointly developed the structure and arguments for the article: NAH and FLWL. Made the critical revisions and approved the final version: NAH, TLD, BLT, and FLWL. All authors reviewed and approved the final article.
Supplementary Materials
Footnotes
Acknowledgments
We would like to thank Aaron DiAntonio's Lab, the Bloomington Drosophila Stock Center, and the Vienna Drosophila RNAi Center for fly stocks. This work benefited from the use of antibodies from the Developmental Studies Hybridoma Bank and Aaron DiAntonio's Lab. We are grateful for the assistance of Carly Gridley with data analysis.