
Abstract
p21-activated kinase 2 (PAK-2) appears to have a dual function in the regulation of cell survival and cell death. Activation of full-length PAK-2 by the p21 G-proteins Rac or Cdc42 stimulates cell survival. However, PAK-2 is unique among the PAK family because it is also activated through proteolytic cleavage by caspase 3 or similar caspases to generate the constitutively active PAK-2p34 fragment. Caspase activation of PAK-2 correlates with the induction of apoptosis in response to many stimuli and recombinant expression of PAK-2p34 has been shown to stimulate apoptosis in several human cell lines. Here, we show that caspase activation of PAK-2 also occurs during cisplatin-induced apoptosis of SH-SY5Y neuroblastoma cells as well as in SH-SY5Y cell culture models for Alzheimer's and Parkinson's disease. Inhibition of mitochondrial complex I or of ubiquitin/proteasome-mediated protein degradation, which both appear to be involved in Parkinson's disease, induce apoptosis and caspase activation of PAK-2 in SH-SY5Y cells. Overexpression of the amyloid precursor protein, which results in accumulation and aggregation of β-amyloid peptide, the main component of β-amyloid plaques in Alzheimer's disease, also induces apoptosis and caspase activation of PAK-2 in SH-SY5Y cells. Expression of the PAK-2 regulatory domain inhibits caspase-activated PAK-2p34 and prevents apoptosis in 293T human embryonic kidney cells, indicating that caspase activation of PAK-2 is directly involved in the apoptotic response. This is the first evidence that caspase activation of PAK-2 correlates with apoptosis in cell culture models of Alzheimer's and Parkinson's disease and that selective inhibition of caspase-activated PAK-2p34 could prevent apoptosis.
Introduction
In a multicellular organism tissue homeostasis is maintained by the coordinated regulation of proliferation, cell survival and programmed cell death. Proliferation, cell survival and programmed cell death are regulated by extracellular signals and intracellular signal transduction pathways. Defects in any component of these signal transduction pathways can trigger abnormalities in cell growth, including cancer and degenerative diseases. Among the signaling molecules that regulate cell survival and cell death is p21-activated kinase 2 (PAK-2). PAK-2 is a ubiquitous member of the PAK family of serine/threonine-specific protein kinases, which in mammals consist of six members; PAK-1 (α-PAK), PAK-2 (γ-PAK) and PAK-3 (β-PAK) form group 1 PAKs, whereas the more distantly related PAK-4, PAK-5 and PAK-6 form group 2 PAKs.1–3 Dysregu-lated PAK signaling has been linked to cancer and neurodegenerative diseases.
PAK activity levels are elevated in several human malignancies. PAK-1 and PAK-2 activity are elevated in human breast cancer cell lines and surgical breast cancer samples.4–7 Elevated PAK activity has been shown to cause loss of contact inhibition, anchorage independent growth, invasion and resistance to anticancer drugs of breast cancer cells.4,5,7,8 Elevated PAK activity also appears to be involved in colorectal cancer, hepatocellular carcinomas and neurofibromatosis type1 and 2.9–13
In contrast reduced or aberrant PAK activity is involved in Parkinson's and Alzheimer's disease. Inhibition of mitochondrial complex I appears to be the central cause of sporadic Parkinson's disease.14,15 It is suggested that inhibition of complex I results in abnormal accumulation and aggregation of α-synuclein, which contributes to neuronal cell death by impairing ubiquitin/proteasome-mediated protein degradation. 14 A functional protein kinase array revealed that aggregated α-synuclein also significantly inhibits PAK-4 activity. 16 Similarly, PAK-1,2,3 activities are markedly reduced or translocated from cytosol to membrane in Alzheimer's disease.17,18 Defects in PAK signaling are accompanied by cofilin pathology and loss of the spine actin-regulatory protein drebrin. β-amyloid peptide (Aβ) was directly involved in PAK signaling defects in Aβ-treated hippocampal neurons and in a transgenic mouse model bearing a mutant amyloid precursor protein (APP) leading to higher Aβ generation. Furthermore, blocking PAK activity with an inhibitory peptide in adult mice caused similar cofilin pathology, loss of drebrin and memory impairment. Importantly, PAK-1,2,3 protein and activity levels have been shown to decline in the hippocampus of moderate and severe Alzheimer's disease subjects. 19 Reduced PAK activity appears to be linked to caspase cleavage of APP at Asp-664. Thirteen month old mice with PDAPP that leads to higher Aβ generation displayed a significant decrease in PAK-1,2,3 activity, which was absent in mice with caspase cleavage-resistant PDAPP-D664A.
PAK-2 is unique among the PAK family; it is cleaved by caspase 3 and to a lesser degree by caspases 8 and 10 to generate the constitutively active PAK-2p34 fragment.20–22 Activated full-length PAK-2 stimulates cell survival whereas the caspase-activated PAK-2p34 fragment induces a cell death response.23–26 Caspase-activated PAK-2p34 but not full-length PAK-2 accumulates in the nucleus and is tightly regulated by ubiquitination and degradation by the proteasome. Surprisingly, expression of histidine (His)-tagged ubiquitin stabilizes PAK-2p34 and as a consequence results in stimulation of cell death. Conjugation of His-tagged ubiquitin to PAK-2p34 prevents further ubiquitin conjugation, and therefore inhibits poly-ubiquitination and degradation by the proteasome. 23 Here, we show that caspase activation of PAK-2 occurs during cisplatin-induced apoptosis of SH-SY5Y human neuroblastoma cells as well as in SH-SY5Y cell culture models of Alzheimer's and Parkinson's disease. In addition, inhibition of the caspase-activated PAK-2p34 by expression of a mutant regulatory domain of PAK-2 appears to prevent cell death.
Material and Methods
Material
The antibody specific for PAK-2 (γPAK-V19), a C-terminal antibody that detects PAK-1, PAK-2 and PAK-3 (αPAK-C19) and the agarose-conjugated Myctag antibody were purchased from Santa Cruz. The antibody for phospho-PAK-2(Thr-402) was obtained from Cell Signaling Technology. The antibody for EGFP Living Colors was from Clontech. The antibody for β-actin, cisplatin, 1-methyl-4-phenyl pyridium (MPP+) and MG-132 were purchased from Sigma. Secondary antibodies conjugated with horseradish peroxidase, SuperSignal West Pico chemilumines-cence reagent and Gelcode Blue Staining Reagent were from Pierce. Restriction enzymes, T4 DNA ligase and cloned Pfu Turbo polymerase were obtained from New England BioLabs. Kits for plasmid DNA isolation were obtained from QIAGEN. TranslT-LTl transfection reagent was from Mirus. Hoechst 33342 was from Molecular Probes. Customized oligonucleotide primers were purchased from Invitrogen. The pMT 107 expression vector for His-tagged ubiquitin was a gift from Dr. D. Bohmann, Rochester University.
Molecular cloning
PAK-2RD-H82A, the PAK-2 regulatory domain mutant that is deficient in binding to Cdc42 or Rac, was generated by site-directed mutagenesis according to the megaprimer PCR method using Pfu Turbo DNA polymerase.27,28 The megaprimer was amplified with H82A-5′ (5′- CCTCTGATTTTGAGGCCACCATC CACG) and RD-3′ (5′- TGAATTCTAGAGGATC CTTACTTAGGGTCACCTATGCT) oligonucleotide primers and wild-type PAK-2 as a template. Then, the megaprimer was used together with PAK-2-5 (5 -TCTGACATATGTCTGATAACGGAGAACTGG) and wild-type PAK-2 as a template to amplify PAK-2RD-H82A, where His-82 was replaced with alanine. PAK-2RD-H82A was subcloned into pExpress/Myc 29 using Ndel and BamHI sites for expression of MycPAK-2RD-H82A. The new cDNA construct was sequenced to confirm the site-directed mutation and ensure the absence of accidental mutations caused by misincorporation during PCR.
Cell culture, adenoviral transduction and DNA transfection
SH-SY5Y human neuroblastoma cells were a gift from Dr. R. Ross, Fordham University, NY, and 293T human embryonic kidney cells were obtained from American Type Culture Collection. SH-SY5Y cells were maintained in DMEM/F12 medium (Invitrogen) supplemented with 10% fetal bovine serum (Hyclone) and penicillin/streptomycin (Invitrogen). 293T cells were maintained in DMEM (Invitrogen) supplemented with 10% fetal bovine serum and penicillin/ streptomycin. Cells were maintained at 37 °C in a humidified atmosphere of 5% CO2. SH-SY5Y cells were differentiated into neuronal cell by incubation in growth medium containing 10 μM retinoic acid for 7 days. Differentiated SH-SY5Y cells were transduced with adenovirus for LacZ, wild-type APP-751 or the familial APP-751-V717F mutant at a MOI of 50, which were gifts from Dr. N. Gottardi-Littell, Northwestern University, Chicago, IL. 293T cells were transfected with plasmid DNA using TranslT-LT1 transfection reagent.
Immunoprecipitation and Western blot
293T cells transfected with EGFP-PAK-2 fusion constructs, Myc-PAK-2RD-H82A and/or His-tagged ubiquitin were lysed in modified RIPA buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.25% deoxycholate 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 50 μM MG-132, 25 mM NaF, 25 mM β-glycerophosphate and 200 μM sodium vanadate). Cleared lysates were prepared by centrifugation for 15 min at 12000 g. Protein concentrations were determined by a Bradford assay using bovine γ-globulin as a protein standard. 500 μg of lysate protein was diluted with PBS to a final concentration of approximately 5 μg/μl and incubated overnight with 20 μl of agarose-conjugated anti-Myc antibody at 4 °C. Immunocomplexes were washed with wash buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM EGTA, 1.5 mM MgCl2, 1% Triton X-100) and analyzed by Western blot. Western blots were performed with cleared lysates (15-30 μg) or immunoprecipitates (from 150 μg cell lysate) by SDS-polyacrylamide gel electrophoresis, transfer onto polyvinylidene fluoride membranes and detection by chemiluminescence using horseradish peroxidase-conjugated secondary antibodies and SuperSignal Pico reagent. X-Ray film exposures were converted into digital images using a Umax scanner and images were processed using Adobe Photoshop 6.0.
Cell death assay
Cells were treated with indicated concentrations of cisplatin, MPP+ or MG-132 for the indicated time periods. Then, cells were stained with 10 μg/ml Hoechst 33342 for 10 min and analyzed by fluorescence microscopy. Images were acquired with a Nikon Eclipse TE2000-U microscope and a Roper Scientific Photometrics CoolSnap ES camera using Metamorph 6.2 software. To determine levels of programmed cell death total cells and cells with apoptotic chro-matin condensation were counted using Metamorph 6.2 software.
Data analysis
Experiments were repeated three times and representative experiments are shown. Two-sided P values were calculated by Fisher's exact test using Graph-Pad Prism 4 software to examine the statistical significance of observed differences in levels of cell death.
Results
Caspase activation of PAK-2 occurs during anti-cancer drug-induced cell death of Neuroblastoma cells
We have shown previously that caspase activation of PAK-2 induces a cell death response and that it occurs during cell death of human breast cancer cells induced with the anti-cancer drug cisplatin.7,23 Cisplatin, which is frequently used in the treatment of neuroblastoma, has been shown to induce apoptosis of SH-SY5Y human neuroblastoma cells. 30 To examine if caspase activation of PAK-2 occurs during cisplatin-induced apoptosis of SH-SY5Y neuroblastoma cells, we treated SH-SY5Y cells with cisplatin and analyzed for apoptotic chromatin condensation and generation of the caspase-activated PAK-2p34 fragment (Fig. 1). Treatment with 20 or 50 μM cisplatin for 24 hours caused high levels of cell death (Fig. 1A) and generation of the caspase-activated PAK-2p34 fragment (Fig. 1B). Treatment with 50 μM cisplatin caused a small but significant increase of cell death at 8 hours and high levels of cell death at 16 and 24 hours (Fig. 1C). Levels of the caspase-activated PAK-2p34 fragment were high at 8 and 16 hours and decreased at 24 hours (Fig. 1D). Levels of β-actin also decreased in late apoptosis indicating the breakdown of all cellular proteins. Therefore, caspase activation of PAK-2 occurs during cisplatin-induced cell death of SH-SY5Y cells.
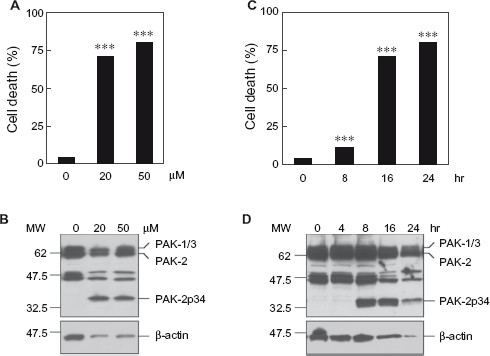
Treatment with the anti-cancer drug cisplatin induces cell death and caspase activation of PAK-2 in SH-SY5Y cells. SH-SY5Y cells were treated with the indicated concentrations of cisplatin for 24 hours (A, B) or for the indicated time points with 50 μM cisplatin (C, D).
Caspase activation of PAK-2 occurs during cell death in a cell culture model of Parkinson's disease
SH-SY5Y cells are frequently used as cell culture models of Parkinson's disease. Treatment with the mitochondrial complex I inhibitor MPP+ is used to mimic cell death in Parkinson's disease.14,15 To determine if caspase activation occurs during MPP+-induced cell death of SH-SY5Y cells, cells were treated with MPP+ and analyzed for apoptotic chromatin condensation and generation of the caspase-activated PAK-2p34 fragment (Fig. 2). Treatment with 2 mM MPP+ for 20 hours caused a small but significant increase of cell death, whereas 5 mM MPP+ caused high levels of cell death (Fig. 2A) and generation of the caspase-activated PAK-2p34 fragment (Fig. 2B). Treatment with 5 mM MPP+ caused increased levels of cell death at 16 hours, which further increased at 20 and 24 hours (Fig. 2C). Levels of the caspase-activated PAK-2p34 fragment were also increased at 16, and further increased at 20 and 24 hours (Fig. 2C). Therefore, caspase activation of PAK-2 occurs during MPP+-induced cell death of SH-SY5Y cells.
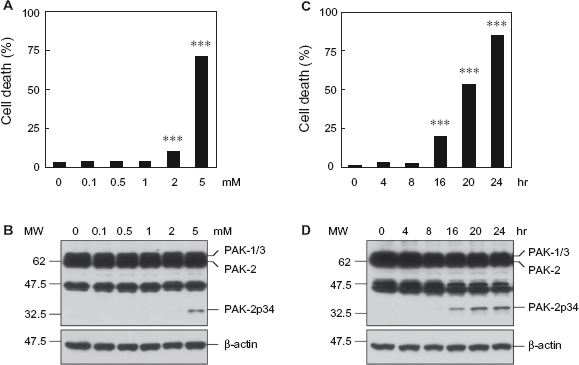
Treatment with the mitochondrial complex I inhibitor MPP+ induces cell death and caspase activation of PAK-2 in SH-SY5Y cells. SH-SY5Y cells were treated with the indicated concentrations of MPP+ for 20 hours (A, B) or for the indicated time points with 5 mM MPP+ (C, D).
Inhibition of mitochondrial complex I with MPP+ results in abnormal accumulation and aggregation of α-synuclein, which contributes to neuronal cell death by impairing ubiquitin/proteasome-mediated protein degradation. 14 To determine if inhibition of the proteasome causes caspase activation of PAK-2 and cell death of SH-SY5Y cells, cells were treated with the proteasome inhibitor MG-132 and analyzed for apoptotic chromatin condensation and generation of the caspase-activated PAK-2p34 fragment (Fig. 3). Treatment with increasing concentrations of MG-132 for 20 hours caused increasing levels of cell death (Fig. 3A). Surprisingly, levels of caspase-activated PAK-2p34 were highest at 1 μM MG-132 and decreased at higher concentrations, suggesting that PAK-2p34 is degraded in late apoptosis (Fig. 3B). Treatment with 5 μM MG132 caused high levels of cell death at 16 hours, which further increased at 20 and 24 hours (Fig. 3C), whereas levels of caspase-activated PAK-2p34 were high at 16 hours but did not increase further at 20 and 24 hours (Fig. 3D). Therefore, caspase-activation of PAK-2 also occurred during MG-132-induced cell death of SH-SY5Y cells.
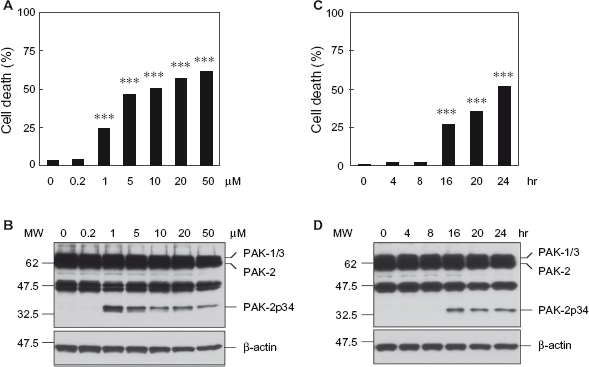
Treatment with the proteasome inhibitor MG-132 induces cell death and caspase activation of PAK-2 in SH-SY5Y cells. SH-SY5Y cells were treated with the indicated concentrations of MG-132 for 20 hours (A, B) or for the indicated time points with 5 μM MG-132 (C, D).
Caspase activation of PAK-2 occurs during cell death in a cell culture model of Alzheimer's disease
SH-SY5Y cells are also frequently used as cell culture models of Alzheimer's disease. Treatment with synthetic Aβ peptides or overexpression of APP are used to mimic Alzheimer's disease.31–33 Overexpression of both wild-type or familial mutants of APP results in increased levels of Aβ peptides. To determine if caspase activation occurs during cell death of SH-SY5Y cells caused by APP overexpression, cells were differentiated with retinoic acid and transduced with adenovirus for wild-type APP-751, the familial APP-751-V717F mutant, or LacZ as a negative control (Fig. 4). Wild-type or mutant APP caused cell death at 48 hours after transduction with adenovirus, which further increased at 72 and 96 hours, whereas LacZ had no effect (Fig. 4 A). Levels of caspase-activated PAK-2p34 were high at 48 hours but decreased at 72 hours in cells with wild-type or mutant APP, whereas LacZ had no effect (Fig. 4B). Caspase-activated PAK-2p34 was also generated when cell death of differentiated SH-SY5Y cells was induced with synthetic Aβ (data not shown). Therefore, caspase-activation of PAK-2 also occurred during Aβ-induced cell death of SH-SY5Y cells.
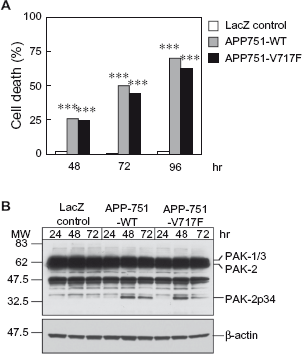
Expression of wild-type or mutant APP induces cell death and caspase-activation of PAK-2 in SH-SY5Y cells.
Inhibition of caspase-activated PAK-2p34 fragment prevents cell death
We have previously shown that caspase-activated PAK-2p34 is ubiquitinated and rapidly degraded by the proteasome, but stabilized by co-expression of His-tagged ubiquitin, which appears to block polyu-biquitination of PAK-2p34. 22 Furthermore, stabilization of PAK-2p34 by co-expression of His-tagged ubiquitin results in cell death. For efficient expression of recombinant proteins we used 293T human embryonic kidney cells in the following studies. The transfection efficiency was 50%-70% as estimated by the number of green fluorescent cells transfected with EGFP-PAK-2 or EGFP-PAK-2T402E (data not shown). Co-expression of Myc-PAK-2RD-H82A, a mutant of the regulatory domain that is deficient in binding to Cdc42 or Rac, inhibited degradation of EGFP-PAK-2p34 and stabilized it to even higher levels than co-expression of His-tagged ubiquitin (Fig. 5A). However, autophosphorylation at Thr-402 in the activation loop, which induces activation, is completely inhibited in the presence of the regulatory domain, indicating that interaction with the regulatory domain inhibits caspase-activated PAK-2p34. Unlike EGFP-PAK-2p34, EGFP-PAK-2 and EGFP-PAK-2T402E, a constitutively active mutant of full-length PAK-2 where Thr-402 has been replaced with glutamic acid, are stable. Since Thr-402 has been replaced with glutamic acid, activity of EGFP-PAK-2T402E cannot be analyzed by autophosphorylation of Thr-402, and autophosphorylation of EGFP-PAK-2 at Thr-402 was not observed. Therefore, interaction of EGFP-PAK-2p34, EGFP-PAK-2 and EGFP-PAK-2T402E with Myc-PAK-2RD-H82A was analyzed by immunoprecipitation with the anti-Myc antibody and Western blot with C-terminal and N-terminal anti-PAK antibodies (Fig. 5B). In contrast to EGFP-PAK-2p34, EGFP-PAK-2 and EGFP-PAK-2T402E did not co-immunoprecipitate with Myc-PAK-2RD-H82A indicating that full-length PAK-2 does not interact with the regulatory domain. EGFP-PAK-2p34 is localized in the nucleus but co-expression of Myc-PAK-2RD-H82A prevents nuclear accumulation of EGFP-PAK-2p34 (Fig. 5C). Therefore, interaction with the regulatory domain also inhibits nuclear localization of caspase-activated PAK-2p34. Since Myc-PAK-2RD-H82A interacts specifically with PAK-2p34 but not full-length PAK-2, we determined if inhibition of caspase-activated PAK-2p34 by expression of Myc-PAK-2RD-H82A prevents cell death. We have shown previously that expression of PAK-2p34 alone does not stimulate cell death of 293T cells and that expression of His-tagged ubiquitin stimulates cell death only slightly from 3 to 10%, but co-expression of PAK-2p34 and His-tagged ubiquitin stimulates cell death to 80%. 23 EGFP-PAK-2p34 and His-tagged ubiquitin were expressed in 293T cells to stabilize EGFP-PAK-2p34 in absence or presence of co-expressed Myc-PAK-2RD-H82A (Fig. 5D). In agreement with our previous results EGFP-PAK-2p34 stabilized by co-expression of His-tagged ubiquitin resulted in ~80% of apoptotic cells. However, co-expression of Myc-PAK-2RD-H82A reduced EGFP-PAK-2p34-induced cell death to 20%. Therefore, inhibition of caspase-activated PAK-2p34 prevents cell death.
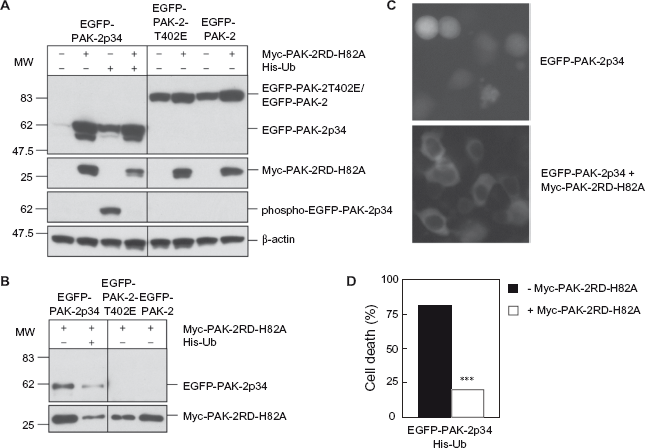
Expression of the PAK-2 regulatory domain inhibits PAK-2p34 activity and PAK-2p34-induced cell death. EGFP-PAK-2p34, EGFP-PAK-2T402E and EGFP-PAK-2 were transfected into 293T cells. Myc-PAK-2RD-H82A and His-tagged ubiquitin (His-Ub) were co-transfected as indicated.
Discussion
We have shown that elevated PAK-2 activity prevents cisplatin-induced apoptosis of breast cancer cells and down-regulates activation of caspase 3 and its own caspase cleavage to the pro-apoptotic PAK-2p34 fragment. 7 In contrast, reduced or aberrant PAK activity appears to be involved in the pathogenesis of neurodegenerative diseases such as Parkinson's and Alzheimer's disease. The ubiquitous PAK-2 isoform is unique among the PAK family because it is also activated by caspase cleavage, which generates the pro-apoptotic PAK-2p34 fragment. The role of caspase activation of PAK-2 in the pathogenesis of Alzheimer's and Parkinson's disease is not known. Here, we investigate if caspase activation of PAK-2 occurs during cisplatin-induced apoptosis of SH-SY5Y neuroblastoma cells as well as in SH-SY5Y cell culture models of Alzheimer's and Parkinson's disease.
SH-SY5Y cells have been shown to undergo apoptosis in response to the DNA damaging anti-cancer drug cisplatin but are resistant to apoptosis in response to taxol, which prevents tubulin depolymerization and mitotic arrest 30 Since we have shown previously that cisplatin-induced apoptosis of human breast cancer cell lines is accompanied by caspase activation of PAK-2, 7 we analyzed apoptosis and caspase activation of PAK-2 in SH-SY5Y cells in response to cisplatin. SH-SY5Y cells showed high levels of apoptosis and caspase-activated PAK-2p34 in response to cisplatin. A previous study has shown that cisplatin induces activation of full-length PAK-2 at 30 and 120 minutes of treatment, but no caspase activation of PAK-2 was observed within the 2-hour treatment. 34 This is consistent with our results that show caspase activation of PAK-2 after 8 hours of cisplatin treatment. Very little apoptosis and caspase-activated PAK-2p34 were observed in response to taxol (data not shown). Therefore, anti-cancer drug-induced apoptosis correlates with caspase activation of PAK-2 and generation of the pro-apoptotic PAK-2p34 fragment.
Loss of anti-apoptotic PAK activity has been associated with Alzheimer's and Parkinson's disease but the role of caspase activation of PAK-2 and generation of the pro-apoptotic PAK-2p34 fragment has not been examined yet. In the present study we show that caspase activation of PAK-2 occurs during MPP+-induced cell death of SH-SY5Y cells, a frequently used cellular model of Parkinson's disease. Since inhibition of mitochondrial complex I is known to result in impaired ubiquitin/proteasome-mediated protein degradation through accumulation and aggregation of α-synuclein, 14 we also examined cell death and caspase activation of PAK-2 in response to the proteasome inhibitor MG-132. We have show previously that MG-132 stimulates caspase activation of PAK-2 in BALB3T3 mouse fibroblasts but have not analyzed levels of cell death. 23 Similar as with MPP+, treatment with the proteasome inhibitor MG-132 results in both caspase activation of PAK-2 and cell death of SH-SY5Y cells. Overexpression of APP results in increased levels of Aβ peptides and is frequently used as a model of neuronal cell death in Alzheimer's disease. Overexpression of wild-type APP-751 or the familial APP-751-V717 mutant in differentiated SH-SY5Y cells results in cell death and caspase activation of PAK-2. Therefore, caspase activation of PAK-2 and generation of the pro-apoptotic PAK-2p34 fragment correlates with cell death in cell culture models of Alzheimer's and Parkinson's disease.
We have shown previously that co-expression of His-tagged ubiquitin prevents poly-ubiquitination and degradation of PAK-2p34 and results in cell death. 22 Here, we show that expression of the regulatory domain of PAK-2 (Myc-PAK-2RD-H82A) also prevents degradation of PAK-2p34 but inhibits stabilized PAK-2p34 and prevents cell death. Since Myc-PAK-2RD-H82A does not bind to full-length PAK-2, it specifically inhibits caspase-activated PAK-2p34. In addition, the Myc-PAK-2RD-H82A mutant of the regulatory domain is defective in binding to Cdc42 and Rac to prevent effects by sequestering these PAK activators. Therefore, caspase-activated PAK-2p34 is actively involved in the cell death response and inhibition of PAK-2p34 prevents cell death. In summary, our results suggest that caspase activation of PAK-2 could be involved in neuronal cell death in Alzheimer's and Parkinson's disease and that selective inhibition of caspase-activated PAK-2p34 could prevent cell death.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and not under consideration by any other publication and has not been published elsewhere. The authors report no conflicts of interest.
Footnotes
Acknowledgements
We thank Dr. R. Ross, Fordham University, NY for the SH-SY5Y human neuroblastoma cells and Dr. N. Gottardi-Littell, Northwestern University, Chicago, IL for the adenoviruses for LacZ, wild-type APP-751 and the familial APP-751-V717F mutant. This work was supported by a grant from the American Parkinson Disease Association, Inc. to RJ.