
Abstract
HIV cure is now the focus of intense research after Timothy Ray Brown (the Berlin patient) set the precedent of being the first and only person cured. A major barrier to achieving this goal on a meaningful scale is an elimination of the latent reservoir, which is thought to comprise CD4-positive cells that harbor integrated, replication-competent HIV provirus. These cells do not express viral proteins, are indistinguishable from uninfected CD4 cells, and are thought to be responsible for HIV viral rebound–-that occurs within weeks of combination anti retroviral therapy (cART) interruption. Modalities to engineer transcriptional stimulation (reactivation) of this dormant integrated HIV provirus, leading to expression of cytotoxic viral proteins, are thought to be a specific way to eradicate the latently infected CD4 pool and are becoming increasingly relevant in the era of HIV cure. HIV protease is one such protein produced after HIV reactivation that cleaves procaspase-8 to generate a novel protein Casp8p41. Casp8p41 then binds to the BH3 domain of BAK, leading to BAK oligomerization, mitochondrial depolarization, and apoptosis. In central memory T cells (TCMs) from HIV-infected patients, an elevated Bcl-2/procaspase-8 ratio was observed, and Casp8p41 binding to Bcl-2 was associated with a lack of reactivation-induced cell death. This was reversed by priming cells with a specific Bcl-2 antagonist prior to reactivation, resulting in increased cell death and decreased HIV DNA in a Casp8p41-dependent pathway. This review describes the biology, clinical relevance, and implications of Casp8p41 for a potential cure.
Introduction to Cell Death and Apoptosis
Cell death is an essential process that allows multicellular organisms to achieve a complex internal organization, maintain homeostasis, and define the boundary between self and the environment. Several viruses have evolved to encode proteins that directly antagonize apoptosis and thus alter the apoptotic phenotype of the infected host cell allowing for latency. HIV is the most clinically relevant example, and latency arises when activated CD4+ T-cells become infected, circumvent death, and subsequently revert to a resting state. The main challenge associated with HIV cure, and one of the key scientific priorities outlined by the International AIDS Society, is to identify interventions that will eradicate the persistent reservoir of latently infected CD4 T-cells. 1 These cells are long lived and thought to be responsible for viral rebound following antiretroviral therapy (cART) interruption. Strategies to eliminate this reservoir will likely involve creative exploitation of existing cell death pathways (Figs. 1 and 2).
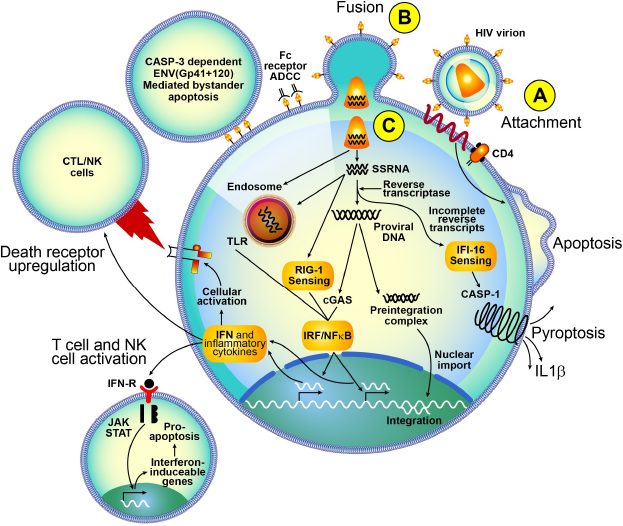
Preintegration cell death in HIV infection. (A) HIV virion attaches to CD4-CCR5 triggering cell death. (B) Fusion of the virion and expression of ENV proteins on the surface of cells may lead to antibody-dependent cytotoxicity and caspase-3-dependent ENV mediated bystander apoptosis. (C) SSRNA and DNA sensed by TLR7, RIG-1, and cGAS mediating INF release, and NF-κB activation. Interferon release may have paracrine apoptotic effects and may activate nearby effector cells (NK cells), while NF-κB leads to cellular activation with upregulation of death receptors. reverse transcription of SSRNA, leading to incomplete reverse transcripts, is sensed by IFI16, leading to casp-1-dependent pyroptosis.
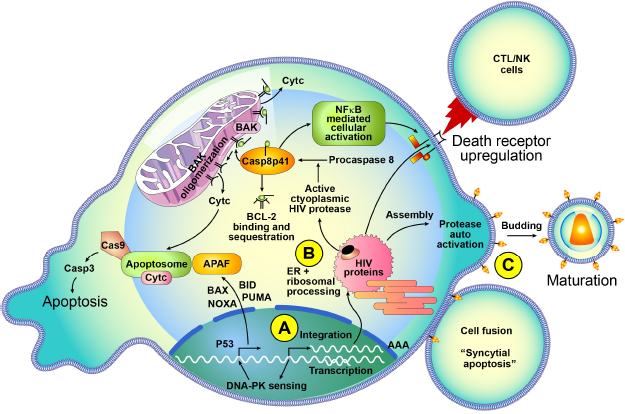
Integration and postintegration cell death in HIV infection. (A) Integration of HIV provirus into human genome is sensed by DNA-PK, leading to P53-mediated apoptosis. (B) Translated HIV proteins undergo ER and ribosomal processing; active cytoplasmic PR cleaves procaspase-8 to Casp8p41, which binds to mitochondrial BAK leading to BAK oligomerization, mitochondrial permeability, and apoptosis. HIV proteins also lead to death receptor upregulation. (C) HIV budding leaves ENV protein on the surface leading to cell fusion and syncytial apoptosis.
Apoptosis
Apoptosis, a form of programmed cell death, can occur by intrinsic or extrinsic pathways (see below) and involves caspase (cysteine-dependent aspartate-specific proteases)-mediated cleavage of cellular proteins, RNA and DNA cleavage by nucleases, nuclear condensation, and formation of membrane-bound fragments called apoptotic bodies. 2 These packaged apoptotic bodies are released and may be phagocytized, thereby circumventing canonical inflammatory cascade(s). 3 The B-cell lymphoma 2 (Bcl-2) family of proteins, with three subtypes: antiapoptotic, proapoptotic multidomain, and Bcl-2 homology domain 3 (BH3) only, together control mitochondrial integrity 4 and regulate the execution phase of apoptosis. 5 During cellular homeostasis, Bax and Bak (proapoptotic Bcl-2 family members) are sequestered by antiapoptotic Bcl-2 family members to preserve mitochondrial membrane integrity. 6 During cellular stress, activated BH3-only proteins also bind to Bcl-2 family members competitively inducing the inhibition of Bax and/or Bak leading to oligomerization of these proteins and loss of mitochondrial integrity. 7 In the setting of HIV infection, relevant to this review, Casp8p41, which is a cleavage product of procaspase-8, directly binds to Bak leading to oligomerization. This loss of membrane potential leads to cessation of adenosine triphosphate (ATP) production in the mitochondria and release of multiple proteins such as cytochrome c (Cyt c), endonuclease G (ENDOG), apoptosis-inducing factor (AIF), direct IAP-binding protein with low protease inhibitors (pI) (DIABLO, also known as second mitochondria-derived activator of caspases [SMAC]), and high-temperature requirement protein A2 (HTRA2). 8 Released Cyt c, apoptotic protease-activating factor 1 (Apaf-1), and ATP then associate to form the “apoptosome”, which activates procaspase-9. Active caspase-9, 9 in turn, activates downstream executioner caspases-3, -6, and -7 to mediate apoptosis.10–12 This sequence of events is conventionally considered an intrinsic apoptotic pathway.
Alternatively, apoptosis may be induced through the extrinsic pathway that occurs when a death-inducing ligand–-Fas ligand (FasL), 13 tumor necrosis factor (TNF), or TNF-related apoptosis-inducing ligand (TRAIL)14,15–-binds to a death signaling receptor (FAS/CD95), TNFR1, or TRAIL receptors 1 or 2, resulting in caspase-8 activation and downstream signaling either to mitochondria or directly to effector caspases. Specifically, caspase-8-mediated cleavage BH3 interacting-domain death agonist (BID) to tBid 16 amplifies the cell death signal by activating the intrinsic apoptotic pathway.
Autophagy
Autophagy is a conserved catabolic pathway by which the cell consumes intracellular components during stress states such as starvation.17,18 Inhibition of autophagy, which is the cell's attempt to survive the stress, may accelerate the dying process. 19 The mammalian target of rapamycin (mTOR), a key regulator of cell growth and differentiation, is also involved in autophagic signaling. 20 When mTOR is inhibited in response to starvation or treatment with rapamycin, the ULK (Unc-51-like kinase)–Atg (autophagy-related gene)–13-FIP200 (FAK family interacting protein of 200 kDa) complex is activated, leading to the sequestration of intracellular components into double-membrane autophagosomes. 21 Beclin-1 is also a protein that stimulates autophagy and is bound to antiapoptotic Bcl-2 family members during cellular homeostasis. 22 During states of cellular stress, Beclin-1 is released and goes on to stimulate autophagy. 23
Necrosis
Necrosis was conventionally thought to be the prototypic unregulated cell death with morphologic traits dissimilar to apoptosis or autophagy. More recently, only severe physical insults such as detergent-induced lysis, complement activation, and hypothermia were found to cause unregulated cell death, while other forms of milder insults trigger a caspase-independent regulated necrotic cell death pathways. 24 Necrotic cell death is characterized by cytoplasmic and organelle swelling, cell membrane rupture, and subsequent inflammation. Scanning electron microscopy shows that this necrotic cell debris is subsequently internalized by macrophages using a macropinocytic mechanism as opposed to apoptotic bodies that are taken up by macrophages with the formation of tight-fitting phagosomes. 3 Pathogen-associated molecular patterns recognized by Toll-like receptors (TLRs), RIG-like receptors (RLRs), and cytosolic NOD-like receptors (NLRs) can also directly trigger necrosis.25–29
Necroptosis
Necroptosis is a regulated form of necrosis triggered by death receptors, independent of caspase activity but dependent on receptor-interacting serine/threonine-protein 1 (RIP1) 30 and/or RIP3. 31 RIP1 is a crucial initiator of death receptor-mediated necrosis, and the term necroptosis was introduced to designate programmed necrosis that depends on RIP1.30,32 Of note, RIP3-dependent, but RIP1-independent, instances of regulated necrosis have recently been identified.33,34 RIP3-dependent necroptosis is insensitive to inhibition by necrostatin 1 (NEC1), which is an RIP1 kinase inhibitor. 35 Necroptosis can be activated through the TNF receptor when caspase-8 is not present, and/or not activated. In those circumstances, TNF receptor type 1-associated death domain (TRADD) mediates activation of RIP1A, leading to necroptotic death, which, unlike apoptosis, is associated with activation of the inflammatory cascade.
Pyroptosis
Pyroptosis is a recently described form of cell death, which is distinct from the other forms described above and is mediated by active caspase-1, resulting in release of pyrogenic interleukin-1β (IL-1β) and IL-18.36,37 It has been shown that in some instances, this is followed by cell death. 38 During pyroptosis, oligomerization of NACHT, LRR, and PYD domains containing protein 1b and 3 (Nalp-1b, Nalp-3), ice protease-activating factor (Ipaf), or absent in melanoma 2 (AIM2) results in the formation of an inflammasome, which in turn activates caspase-1. A noncanonical inflammasome pathway triggered by lipopolysaccharide (LPS) has also been recently described. This involves activation of caspase-11 leading to cell lysis that differs from pyroptosis in that IL-1β and IL-18 are not secreted in the absence of caspase-1 activation. 39 Gasdermin D, a component of the inflammasome, is cleaved by caspase-1 during inflammasome activation, and the N-terminal cleavage product is capable of inducing cell death. This cleavage event is required for the release of IL-1b and in a gasdermin D knockout cell line apoptosis, rather than pyroptosis became more apparent on exposure to the appropriate toxic stimulus. 40 Alternatively, ASC dimers (apoptosis-associated speck protein containing a caspase activation and recruitment domain) may associate to form a pyroptosome, resulting in caspase-1 activation. 41
Other Forms of Cell Death
There are other modes of cell death that have been described and studied, yet because they have not been associated with HIV, we will mention but not explain them in detail. They include the following: mitotic catastrophe that is triggered by aberrant mitosis, 42 anoikis that is death restricted to adherent cells,43,44 entosis that is characterized by a “cell-in-cell” morphotype in nonphagocytic cells, parthanatos that constitutes a caspase-independent cell death pathway, 45 and netosis that is mediated by neutrophil extracellular traps (NETs), which are released by neutrophils and eosinophils. 41
Cell Death and HIV
Causes of CD4 T-cell death that occurs during HIV infection are multifactorial and involve death of both HIV-infected and HIV-uninfected cells. Infected cell death refers to the death of cells that contain HIV genetic sequences–-either early following infection when HIV RNA is reverse transcribed into DNA or later in the viral life cycle, after HIV DNA has integrated into the host genome and is replicating progeny virions. Uninfected cell death refers to death of cells that do not contain HIV genetic material, yet this bystander cell death can occur due to the effects of soluble HIV proteins, as well as other immune mechanisms. Although studies have implicated autophagy, 46 necrosis, 47 necroptosis, 48 pyroptosis, 37 and apoptosis in HIV-induced cell death, the relative contribution of each form is unknown and further research into this is a key priority. 49 Furthermore, it is unknown if unique forms of cell death predominate in certain tissues/microenvironments, although it is accepted that apoptosis contributes significantly to loss of bystander CD4 cells in the setting of HIV infection.50–53
Infected cell death
Infected cell death can be triggered at a preintegration stage or at an integration/postintegration stage and depends on different sensors in different cell types. Recent studies in human lymphoid aggregate cultures show that cell death is related to the degree of cellular activation, and only a small percent of activated cells die via caspase-3-dependent apoptosis in the setting of acute HIV infections. About 95% of these cells are abortively infected with the accumulation of intracellular incomplete viral DNA transcripts. Interferon-γ-inducible protein 16 (IFI16) senses these transcripts and leads to inflammasome assembly, caspase-1 activation, and subsequent cell death by pyroptosis. 37 Cytosolic HIV DNA is also sensed by cyclic GMP-AMP synthase (cGAS), leading to the production of cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), which binds the stimulator of interferon genes (STING), resulting in interferon-β production via interferon regulatory factor 3 (IRF3). It may also occur at the time of HIV DNA integration into the host genome causing activation of DNA-dependent protein kinase (DNA-PK), which causes phosphorylation of p53 and histone H2AX. 54 Immune sensing of HIV in an infected cell is a crucial prelude to death. In plasmacytoid dendritic cells (pDCs), TLR7-sensing of the HIV RNA produces IFN-a.55–57 Infection in monocyte-derived dendritic cells is restricted by SAMHD1, which depletes the cellular pool of nucleotides required for HIV-1 reverse transcription. In these monocyte-derived dendritic cells, HIV-1 with vpx (to overcome restriction by SAMHD1) or HIV-2 DNA are sensed by the cytosolic DNA sensor cyclic GMP-AMP synthase (cGAS), and this is dependent on capsid–CypA interaction. 58
Uninfected cell death
Apoptosis is one of the major mechanisms of CD4+ T-cell depletion that can be observed in the HIV-negative cell fraction. 53 Several triggers for this “bystander death” have been implicated and include overexpression of death ligands (FasL, TNF, and TRAIL)59,60 on immune cells, direct cytotoxicity of a number of soluble HIV proteins (eg, Gp120, Tat, Nef, Vpr),61–64 and increased rates of activation-induced cell death (AICD), 65 which is a biological failsafe mechanism to eliminate active T-cells after the termination of an immune response.66,67 HIV infection is also associated with abnormalities in the thymus 68 and reduction of input of naïve CD4+ T-cells into the peripheral pool. 69 Cellular fusion leading to a cytoplasmic mass referred to as syncytia has been observed between HIV-infected and HIV-uninfected cells. Syncytial apoptosis that may be mediated by p53 is triggered by the fusion of HIV-infected cells expressing HIV Env (gp120/gp41) with uninfected target cells, resulting in syncytia formation and apoptosis.70,71
HIV Proteins and Cell Death
Gag, Pol, and Env are structural polyproteins cleaved by the action of HIV protease (PR) into: (1) Gag proteins include MA (matrix), CA (capsid), NC (nucleocapsid), and p6, (2) Env proteins include gp120 and gp41, and (3) Pol proteins include PR, reverse transcriptase (RT), and integrase (IN). 72 HIV encodes six additional proteins: the regulatory proteins Tat and Rev and the four accessory proteins Vif, Vpr, Nef, and Vpu. 73 Several of these proteins can trigger cell death. Relevant to the discussion below, Tat has been shown to increase intracellular caspase-8, 74 while other proteins such as Vpu, 75 Nef, 76 and gp120 77 increase susceptibility to extrinsic apoptosis. Vpr has also been shown to bind to mitochondrial proteins like Bax and ANT, leading to Cyt c release and subsequent apoptosis.78,79
HIV Protease
Intracellularly, HIV-1 PR is expressed as part of the Gag-Pol polyprotein precursor: Gag will be processed to viral structural proteins, while Pol is eventually processed to viral PR, RT, and IN. 72 Gag and Gag-Pol along with other viral proteins and HIV RNA then bud into immature HIV-1 particles, and this process is thought to coincide with the self-activation of PR. Despite this widely accepted dogma, more recent studies have demonstrated the activity of HIV PR in the cytoplasm as evident by the intracellular accumulation of appropriately processed viral proteins in HIV-infected cells. 80 Also, we have directly measured PR function in the cytosolic compartment and found it to be as robust as the membrane fraction that contains immature virions. 80 It is known that HIV PR-transfected cells die, and a positive correlation has also been shown between the degree of cytoplasmic processing and cell lysis. 81 The substrate specificity of HIV PR is degenerate and extends to cellular proteins like Bcl-2, 82 actin, 83 desmon, eukaryotic initiation factor 4G, 84 vimentin, a subunit of eukaryotic translation factor 3 (eIF3d), 85 laminin B, prointerleukin-1, and procaspase-8.82,83 Of these, procaspase-8 is of particular importance as its absence abrogates HIV PR-induced mitochondrial depolarization and nuclear fragmentation. 80
Casp8p41 Biology
Although HIV PR induces CD4 cell apoptosis, 83 coincubation of nuclei with HIV-1 PR does not induce apoptotic nuclear changes, 86 suggesting the involvement of an intermediate cytoplasmic factor(s) that may be acted upon by HIV-1 PR. Using a cell-free system, we observed that following the addition of PR to cytosolic extracts, the earliest protein to be processed was procaspase-8, which occurred before loss of mitochondrial outer membrane permeabilization (MOMP), Cyt c release, caspase 9 and 3 activation, and nuclear fragmentation. Even more importantly, cytosolic extracts from cells lacking procaspase-8 expression did not undergo apoptotic changes. 80
In vitro digestion of procaspase-8 engineered with a carboxyl-terminal Myc epitope and an amino-terminal Flag epitope with HIV PR allowed identification of the HIV PR cleavage site between Phe355 and Phe356. When these cleavage sites were mutated in full-length procaspase-8 (F355R, F356N), incubation with HIV-PR resulted in negligible cleavage, 87 while incubation of wild-type (WT) procaspase-8 with HIV-1 PR resulted in a p41 procaspase-8 fragment (Casp8p41). 87
Casp8p41 Kills Cells and Mediates Inflammation
The novel discovery of HIV PR-cleaving procaspase-8 led to further studies to understand the biological function of the cleavage fragment Casp8p41. When we questioned if the mechanism of HIV PR cytotoxicity was due to Casp8p41, we observed that their respective death pathways were identical in both cell-free systems 88 and in primary CD4 T-cells transfected with GFP-Casp8p41. In both instances, Casp8p41 expression induced terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) positivity, active caspase 3 expression, and annexin positivity in a time-dependent manner. 87 Somewhat unexpectedly, Casp8p41 expression was also shown to enhance HIV replication by activating NF-κB-dependent HIV long terminal repeat, 89 an observation that can be rationalized by the obligate requirement for caspase-8 in maximizing T-cell activation. 90 Consistent with a role of Casp8p41 in T-cell activation, Casp8p41expression independently promotes proinflammatory cytokine production and release, 89 which is characteristic of untreated HIV infection.
Mechanism of Casp8p41-mediated Cell Death
Cleavage of procaspase-8 by HIV PR as mentioned above results in two fragments, one of 41 kd, which contains two death-effector domains (DEDs) but lacks the catalytic cysteine of an active caspase (Casp8p41), and another of 14 kd, which contains an active site but no DED that is degraded and is not detected by autoradiography. To understand how Casp8p41 induces cell death despite lacking an active catalytic cysteine, an unbiased screen using lentiviral-expressed short hairpin RNA (shRNAs) was used to identify genes that diminish Casp8p41 killing. Because GFP-Casp8p41 induces death within 24 hours, any cell that survived for 48 hours following Casp8p41 expression must be altered in such a way as to confer resistance to Casp8p41-mediated killing. A total of 1558 of 27,000 shRNAs conferred partial resistance to Casp8p41 killing, including caspase-9 and Bak, 91 which were further validated by independent knockdown experiments confirming their role in Casp8p41-induced death. 91 Altogether, these findings suggest that Casp8p41 translocates to the mitochondria to release Cyt c and raises the possibility of a direct interaction between Bak and Casp8p41. Using surface plasmon resonance (SPR) and reciprocal immunoprecipitation, we successfully identified an interaction between Casp8p41 and Bak. The site of interaction was mapped to a BH3-like domain in Casp8p41, which is unmasked by the PR cleavage event binding to a BH3-binding groove in Bak. This binding leads to Bak oligomerization, loss of MOMP, and cell death. 91 These observations provide new insights into the mechanism of T-cell death during productive HIV infection.
Despite our understanding of the role of Casp8p41 in the intrinsic apoptotic mechanism of cellular death, it remained unexplained why TCMs do not consistently die, despite production of Casp8p41 during viral reactivation. These cells were observed to have a high Bcl-2/procaspase-8 ratio, and this was thought to render the cell relatively resistant to apoptosis. Based on our previous experiments showing that Casp8p41 binds to the BH3 groove of BAK, we found that Casp8p41 was also bound and sequestered by Bcl-2 in these cells with high avidity, thereby circumventing apoptosis. Bcl-2 overexpression was also shown to decrease cell death induced by Casp8p41 and increase viral replication in Jurkat T-cells stably overexpressing BCL-2 that were transfected with GFP-Casp8p41. Cell death and consequently decrease in HIV proviral DNA that is a marker for latency was seen in TCMs from HIV-infected patients on cART when they were reactivated with CD3/CD28 in the presence of a selective Bcl-2 antagonist venetoclax. 92
Clinical Implications
We have also assessed the clinical implications of this novel protein using a monoclonal antibody generated by immunizing mice with the c-terminal 11 amino acids of Casp8p41 and screened for antibodies that identified the c-terminal 3 amino acids, but not full-length caspase-8, nor Casp8p43 (a processing intermediate that is generated during death receptor-initiated caspase-8-dependent apoptosis). 93 This reagent allowed us to determine that Casp8p41 is a relatively specific marker of HIV-1 PR-induced caspase-8 processing and subsequent apoptosis. 87 Next, we assessed expression of Casp8p41 in cells from HIV-1-infected patients; intracellular staining with the anti-Casp8p41 antibody confirmed specificity to HIV-infected versus HIV-uninfected cells, and apoptotic versus nonapoptotic cells. Absent in peripheral blood lymphocytes (PBLs) from HIV-uninfected patients, Casp8p41 is expressed in < 1% of PBLs from HIV-infected subjects, consistent with the frequency of circulating infected cells, and is expressed most frequently in central memory CD4 T-cells. Furthermore, Casp8p41 expression is higher in patients with active viremia compared to patients on suppressive anti-retroviral therapy (ART). 94
HIV disease progression has been associated with several factors, including clinical presentation of primary HIV seroconversion, 95 social and psychosocial factors, 96 state of immune activation,97,98 HIV viral load, 99 baseline CD4 count, 99 activated CD8 T-cell count, 100 and bacterial 16s DNA levels. 101 In addition, Casp8p41 content is inversely correlated with CD4 T-cell count, and change in Casp8p41 content is associated with absolute CD4 T-cell change over time; thus, patients with high Casp8p41 levels will lose CD4 cells, whereas patients with low or undetectable Casp8p41 levels will not. Change in Casp8p41 level was shown to be a better predictor of CD4 T-cell count trend over time than other indicators like activated CD8 T-cell percentage or viral load and was comparable to bacterial 16s DNA levels. 94 Casp8p41 content in memory CD4 T-cells was inversely correlated with absolute CD4 count while the odds ratio for an increase in CD4 T-cell count after a decrease in Casp8p41 expression was 5.27. 94 Typically, in patients who initiate effective cART, an improvement in the CD4 cell count coincides with a decrease of HIV viral load to undetectable levels. 102 Patients who have an improvement in CD4 count, but fail to suppress viral replication, are referred to as discordant responders. Such discordance occurs in about 10%-20% of patients initiating therapy (typically PI-based), and these patients have reduced rates of apoptosis of CD4 T-cells103,104 and higher T-cell receptor excision circle (TREC) content. 105 We have studied these discordant patients and identified an increased prevalence of the PR mutations I54V and V82A. In vitro recombinant I54V or V82A HIV PRs cleave procaspase-8 less efficiently than WT PR (with unimpaired cleaving of gag/pol). Moreover, recombinant HIV virions containing I54V or V82A HIV PRs generate less Casp8p41 in primary CD4 T-cells following infection, resulting in reduced infected cell death. 106 An association between Casp8p41 content in memory CD4 cells and CD4 cell decline while on cART was also observed in a cohort of patients from the SMART (Strategies for Management of Antiretroviral Therapy) study. 107 Ongoing production of Casp8p41 in these patients confers an increased risk of CD4 T-cell losses, suggesting that subclinical HIV replication is driving Casp8p41, which in turn causes a CD4+ T-cell decline.
Future Directions
Advances in the understanding of molecular mechanisms by which Casp8p41 initiates cell death may lead to novel strategies aimed at reducing HIV-induced cell death, and thus, inhibitors of Casp8p41 may have use in decreasing the CD4 decline seen in HIV infection. Alternatively, one of the main challenges facing HIV cure research is eliminating the latent reservoir, and the observed paradigm is that reactivation does not kill these cells. One explanation for this observation is that latent cells downregulate procaspase-8, become apoptosis resistant, 108 and render the Casp8p41 pathway of cell death ineffective. Strategies to sensitize cells that harbor latent HIV toward a proapoptotic phenotype by increasing procaspase-8 expression and then inducing viral reactivation with available agents could lead to enhanced death of these cells and uncover new HIV “prime, shock, and kill” cure strategies.
Footnotes
Abbreviations
MOMP, mitochondrial outer membrane permeabilization; AIF, apoptosis-inducing factor; ENDOG, endonuclease G; DIABLO, direct IAP-binding protein with low pI; SMAC, second mitochondria-derived activator of caspases; HTRA2, high-temperature requirement protein A2; Cyt c, cytochrome c; Apaf-1, apoptotic protease-activating factor 1; TNF, tumor necrosis factor; FasL, Fas ligand; BID, BH3 interacting-domain death agonist; mTOR, mammalian target of rapamycin; ULK, Unc-51-like kinase; Atg, autophagy-related gene; TLRs, Toll-like receptors; NLR, NOD-like receptors; RLRs, RIG-I-like receptors; RIP1, receptor-interacting serine/threonine-protein 1; NEC1, necrostatin 1; TRADD, tumor necrosis factor receptor type 1-associated DEATH domain protein; Nalp, NACHT, LRR, and PYD domains containing protein; Ipaf, Ice protease-activating factor; AICD, activation-induced cell death; AIM2, absent in melanoma 2; ASC, apoptosis-associated speck protein containing a caspase activation and recruitment domain; NETs, neutrophil extracellular traps; IFI16, interferon-γ-inducible protein 16; DNA-PK, DNA-dependent protein kinase; pDCs, plasmacytoid dendritic cells; PR, protease.
Author Contributions
Conceived and designed the experiments: ADB, NWC. Analyzed the data: RS, ADB, NWC. Wrote the first draft of the manuscript: RS, ADB, NWC. Contributed to the writing of the manuscript: RS, ADB, NWC. Agree with manuscript results and conclusions: RS, ADB, NWC. Jointly developed the structure and arguments for the paper: RS, ADB, NWC. Made critical revisions and approved final version: RS, ADB, NWC. All authors reviewed and approved of the final manuscript.