
Abstract
7-Oxysterols are major toxic components in oxidized low-density lipoprotein and human atheroma lesions, which cause lysosomal membrane permeabilization (LMP) and cell death. Autophagy may function as a survival mechanism in this process. Here, we investigated whether 7-oxysterols mixed in an atheroma-relevant proportion induce autophagy, whether autophagy induction influences 7-oxysterol-mediated cell death, and the underlying mechanisms, by focusing on cellular lipid levels, oxidative stress, and LMP in 7-oxysterol-treated macrophages. We found that 7-oxysterols induced cellular lipid accumulation, autophagy dysfunction, and cell death in the form of both apoptosis and necrosis. Exposure to 7-oxysterols induced autophagic vacuole synthesis in the form of increased autophagy marker microtubule-associated protein 1A/1B-light chain 3 (LC3) and LC3-phosphatidylethanolamine conjugate (LC3-II) and autophagic vacuole formation. This led to an accumulation of p62, indicating a reduction in autophagic vacuole degradation. Importantly, autophagy induction significantly reduced 7-oxysterol-mediated cell death by diminishing LMP and oxidative stress. Moreover, autophagy induction minimized cellular lipid accumulation induced by 7-oxysterols. These findings highlight the importance of autophagy in combating cellular stress, LMP, and cell death in atherosclerosis. Therefore, activation of the autophagy pathway may be a potential therapeutic strategy for prevention of necrotic core formation in atherosclerotic lesions.
Introduction
Apoptotic cell death occurring in atheroma plaques contributes to plaque development, plaque rupture, and atherothrombosis. 1 The apoptosis of macrophages, among several other cell types, is of significant importance for the stability of advanced plaques. Among various pro-apoptotic stimuli, 7-oxysterols, the major toxic compounds found in oxidized low-density lipoprotein and atherosclerotic lesions, are of great significance. We have previously reported that 7-oxysterol-mediated cell death is associated with lysosomal membrane permeabilization (LMP) and increased levels of cellular reactive oxygen species (ROS).2,3 The accumulation of oxidized lipids, including oxysterols, in cells induces the formation of autophagy vacuoles.4–8
Autophagy is a lysosomal degradation pathway for transport of cytoplasmic material and is activated under cellular stress conditions. Autophagy can promote cell survival by the elimination of damaged organelles and protein aggregates to generate free amino acids and fatty acids to maintain cellular function. However, it can also promote cell death through excessive self-digestion and degradation of essential cellular constituents.
9
A total of 31 autophagy-related (Atg) proteins have been identified in yeast. Among the 31 Atg proteins, 18 Atg proteins, including Atg5, are involved in autophagic vacuole formation. LC3, a mammalian homolog of Atg8, has been identified on the autophagosomal inner membrane and has been proposed to function as a receptor for a selective substrate, p62/Sequestosome 1 (
In the present study, we investigated whether 7-oxysterols mixed in an atheroma-relevant proportion induces autophagy, whether autophagy induction influences 7-oxysterol-mediated apoptosis, and the underlying mechanisms, by focusing on cellular lipid levels, oxidative stress, and LMP in 7-oxysterol-treated-a human leukemic cell line (THP-1) cells. These cells have been widely used in cardiovascular studies, as they resemble primary monocytes/macrophages isolated from healthy donors and from patients with diabetes mellitus. The cells also mimic the in situ alteration of macrophages in the adipose tissue of obese subjects and in atherosclerotic lesions. 14
Materials and Methods
Cell culture and experimental procedure
Human monocyctic THP-1 cells (ATCC) were maintained in Roswell Park Memorial Institute-1640 culture medium supplemented with 10% fetal bovine serum in a humidified atmosphere (5% CO2) at 37°C and sub-cultivated twice a week. In some experiments, THP-1 cells were differentiated into macrophages after incubation with phorbol myristate acetate (300 nM) for 24 hours. After washing with the culture medium, the cells were further cultured for 24 hours under standard culture conditions before the initiation of the experiment. Mouse embryonic fibroblasts (MEF) lacking Atg5 gene expression (Atg5−/− MEF) or MEF with Atg5 gene expression (Atg5+/+ MEF) (Cell Bank) were cultured in Dulbecco's Modified Eagle's Medium supplemented with 10% fetal bovine serum in a humidified atmosphere (5% CO2) at 37°C and sub-cultivated twice a week.
For experiments, the abovementioned cells (seeded at 3 × 105 cells/ml) were treated with a mixture of 7β-hydroxycholesterol (7βOH) and 7keto-cholesterol (7keto) (Sigma-Aldrich) at a ratio of 1:1.8 (28 μM; referred to as 2mix) for 12, 24, or 48 hours as described previously. 3 Cells treated with cholesterol or ethanol were used as controls. In some experiments, cells were pretreated for 1 hour with the autophagy inducer, rapamycin (1 μM, Sigma-Aldrich), or the autophagy inhibitor, 3-methyladenine (3MA; 1 mM, Sigma), or the lysosomotropic agent, chloroquine (CQ; 40 μM, Sigma-Aldrich), and then exposed to 2mix for an additional 6 hours to 24 hours in the presence of either the inducer or the inhibitor. After the treatment, the cells were collected and analyzed as described below.
Lysosomal membrane integrity
The integrity of the lysosomal membranes was assessed using lysotracker (Molecular Probes) staining or acridine orange (AO) relocation test, as established previously. 15 In brief, after different treatments, cells were collected, stained with lysotracker (75 nM for 25 minutes at 37°C), and analyzed using flow cytometry (BD Biosciences), and the flow cytometric data were analyzed by using CellQuest software (BD Biosciences). Cells with decreased lysotracker red fluorescence intensity, below the basal levels of the lysotracker peak in histograms, were gated and identified as cells with LMP. For the AO relocation test, cells were first stained with AO and then were subjected to different treatments for different times, and analyzed by confocal microscopy. Cells with increased cytosolic/nuclear AO green fluorescence were identified as cells with LMP. LMP was also assessed by cathepsin D immunocytochemistry. In brief, after different treatments, cells were collected, fixed with 2% paraformaldehyde (PFA), permeabilized with 0.1% saponin in phosphate-buffered saline (PBS), incubated with anti-cathepsin D antibody (R&D Systems), and analyzed using confocal microscopy.
Reactive oxygen species
Intracellular reactive oxygen species (ROS) were assessed using flow cytometry following dihydroethidium (DHE; Molecular Probes) staining. Cells were collected, washed once with PBS, incubated for 15 minutes at 37°C with 10 μM DHE, and analyzed using flow cytometry. Cells with increased DHE red fluorescence, above the basal levels of the DHE peak in histograms, were gated and identified as cells with ROS.
Apoptosis and necrosis
The amount of cell death was assayed by detection of phosphatidylserine exposure using flow cytometry following Annexin V (AV)/propidium iodide (PI) (Roche Mannheim) staining. Briefly, control and treated cells were collected, washed with PBS, and stained for 20 minutes on ice with AV/PI. Cells were scored as early apoptotic when they were predominantly AV positive, while cells positive for both AV and PI were scored as late apoptotic or post-apoptotic necrotic. The percentages of both early and late apoptotic cells were analyzed to determine the amount of cell death. In some experiments, apoptotic cells were detected by immunocytochemistry using an antibody-recognizing activated caspase-3 (BD PharMingen).
Intracellular lipid levels
Cellular lipid levels were assayed by oil red O (Sigma-Aldrich) staining. Briefly, cells were fixed with 2% formalin, stained for 6 minutes with 0.15% oil red O in 76% methanol and 0.2 M sodium hydroxide, washed with water, counterstained with hematoxylin, and visualized by light microscopy.
Flow cytometry and immunocytochemistry
Autophagy was assayed by immunocytochemistry of LC3β, Atg5, or p62/
Western blot analysis
Whole cell lysate proteins were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (15% T) on a mini-protean II electrophoresis cell (Bio-Rad Laboratories Inc.). The separated proteins were then transferred to a polyvinylidene fluoride membrane (BioRad Laboratories Inc.). The membranes were blocked with 5% non-fat dried milk in Tris-Buffered Saline (TBS) and then incubated overnight with anti-LC3 (1:1,000) in mixture of Tris-Buffered Saline and Tween 20 (TTBS) with 2% nonfat dried milk at 4°C. Then, the membranes were incubated with horse radish peroxidase-conjugated secondary antibodies (1:1,000; 22°C; 60 minutes); the antigen/antibody conjugate was detected using chemiluminescence enhanced chemiluminescence (ECL) solution (GE Healthcare) and visualized using a cooled charged coupled device camera digitizing at a resolution of 1,340 × 1,040 pixels (VersaDoc™ Imaging System; Bio-Rad Laboratories Inc.). α-Actin was blotted on the same stripped membrane as an internal control for loading.
Transmission electron microscopy (TEM)
TEM was used to assess the number of autophagic vacuoles in THP-1 cells. The TEM technique was performed in the same manner as described previously. 15 Cells were randomly scanned from each group and the numbers of autophagic vacuoles in each cell were counted.
Statistics
For statistical analyses, one-way analysis of variance followed by a post hoc Newman–Keuls test was used for multiple comparisons. Results are given as mean ± SEM, where
Results
7-Oxysterols induce lipid accumulation and dysfunction of autophagy
We first assessed whether cellular lipid accumulation is related to autophagy. THP-1 cells were treated with 7-oxysterols (2mix) and cellular lipids were assayed by oil red O staining. Untreated cells showed fewer lipid droplets in the cytoplasm and some cells showed mitosis morphology (arrows), while after 12-hour exposure to 2mix, many cells exhibited lipid accumulation in the cytoplasm, and after 24-hour exposure, lipid accumulation was observed in almost all cells (Fig. 1A). Many of these cells were shrunken with nuclear condensation (Fig. 1A, arrowheads). We then examined the expression of several autophagy proteins (LC3β, Atg5, and p62/
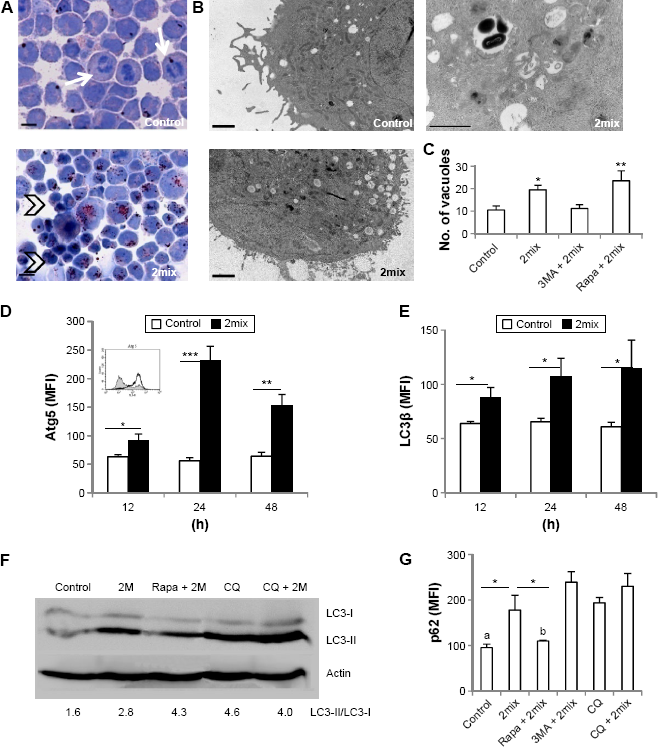
Exposure to a mixture of 7-oxysterols (2mix) causes lipid accumulation and autophagy dysfunction. THP-1 monocytes or THP-1 differentiated macrophages were treated with or without 2mix for 12 hours to 48 hours. In some experiments, cells were pretreated with the autophagy inhibitor 3MA or the autophagy inducer rapamycin for 1 hour and then exposed to 2mix or treated with the lysosomatropic agent CQ for 24 hours or CQ for 1 hour and 2mix for 24 hours. (A) Oil red O-stained THP-1 monocytes counterstained with hematoxylin (24 hours), bars = 100 μm. (B and C) Accumulation of autophagy vacuoles in THP-1 differentiated macrophages assayed by TEM and quantified by image analysis. (B) Photographs of low power view of control and 2mix-treated cells (two photographs on the left-hand side), bars = 2 μm or a higher magnified photograph (photograph on the top right-hand side) demonstrates autophagic vacuoles with double-membrane structure containing undigested cytoplasmic materials, bar = 1 μm. (C) Quantification of numbers of autophagic vacuoles, *
Autophagy induction minimizes cell death and lipid accumulation in 7-oxysterol-exposed cells
To investigate whether autophagy induction or inhibition influences cell death induced by 7-oxysterols, THP-1 cells were pretreated with the autophagy inhibitor 3MA or the autophagy inducer rapamycin. The 2mix treatment induced a time-dependent cell death response, in the form of both apoptosis and necrosis, which was significantly enhanced by 3MA (Fig. 2A) and significantly reduced by rapamycin, even up to 48 hours (Fig. 2B). 3MA treatment alone did not show any cytotoxic effect up to 48 hours. The genetic loss of Atg5 function in 7-oxysterol-mediated cell death was further investigated in Atg5-knockout MEF. Following 7keto treatment, Atg5−/−cells were apoptotic and were characterized by cell shrinkage and nuclear condensation, which occurred earlier (<14 hours) than that in Atg5+/+ cells. Moreover, cell numbers after exposure to 7keto were pronouncedly reduced at 24 hours in Atg5−/− cells (43 ± 4.01) when compared to Atg5+/+ cells (112 ± 10.7), further suggesting a cytoprotective role of Atg5 in 7-oxysterol-induced cell death.
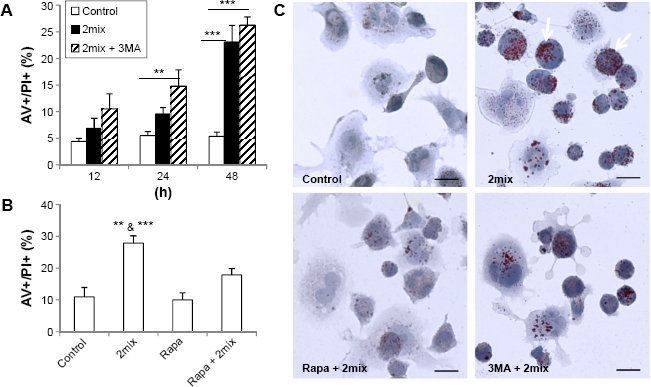
Up- and downregulation of autophagy regulates cellular lipid accumulation and cell death in 7-oxysterol-treated cells. (A and B) THP-1 monocytes were treated for different time periods (A) or for 48 hours (B) with 2mix or pretreated with 3MA (A) or rapamycin (B) for 1 hour and then exposed to 2mix. The cells were then stained with AV/PI and analyzed using flow cytometry [
In order to examine whether autophagy can alter the levels of 2mix-induced lipid accumulation, THP-1 differentiated macrophages were treated with rapamycin or 3MA together with 2mix for 24 hours. Cellular lipids were assayed by oil red O, and nuclear/cell morphology was analyzed by hematoxylin staining. As shown in Figure 2C, lipid accumulation was induced by the exposure to 2mix (arrows) and was decreased in the presence of the autophagy inducer rapamycin, while the autophagy inhibitor 3MA had no effect. In summary, these results indicate that autophagy induction protects against cell death induced by 7-oxysterols and lipid accumulation.
Cytoprotective autophagy regulates levels of ROS and LMP in 7-oxysterol-exposed cells
ROS, as multifaceted signaling molecules, have been implicated in apoptosis and autophagy. We have earlier demonstrated that 7-oxysterols induce the formation of ROS in monocytes and macrophages. 3 To investigate if ROS formation is involved in the cytoprotective effect induced by autophagy, we analyzed cellular levels of ROS production in response to autophagy inhibition by 3MA or induction by rapamycin in 2mix-exposed THP-1 cells. When compared to control cells, 2mix induced time-dependent production of ROS, which was further enhanced by 3MA at 24 hours (Fig. 3A). Moreover, the exposure to rapamycin resulted in significant reduction of cellular ROS in 2mix-treated cells (Fig. 3B).
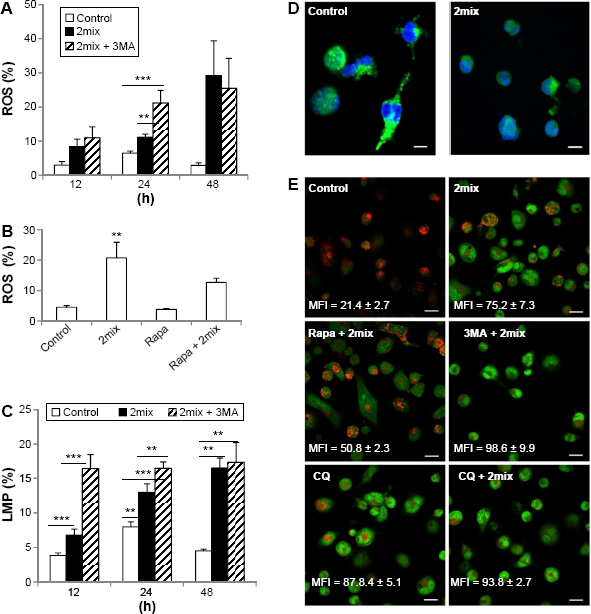
Up- and downregulation of autophagy regulates oxidative stress and LMP induced by 7-oxysterols. THP-1 cells were treated with 2mix for different time periods (A and C) or for 48 hours (B) or for 12 hours (D) or pre-treated with 3MA (A) or rapamycin (B) for 1 hour and then exposed to 2mix. The cells were then collected, stained with DHE for ROS (A and B) with lysotracker (C) or immunostained for cathepsin D (D) for LMP. In some experiments (E), LMP was monitored by AO relocation test. In this setting, THP-1 differentiated macrophages were stained with AO first and then subjected to different treatments for 6 hours, followed by fluorescence microscopy. (A) Autophagy inhibition by 3MA enhances cellular ROS induced by 2mix (
Our previous studies suggested that 7-oxysterols cause apoptotic cell death through lysosomal pathways and the induction of cellular stress.2,3 In this study, the effects of autophagy inhibition by 3MA or induction by rapamycin in the 2mix-induced LMP were analyzed by flow cytometry after lysotracker staining or by fluorescence microscopy following cathepsin D immunocytochemistry or AO relocation test. As shown in Figure 3C, 2mix induced a time-dependent LMP, which was statistically significant after 24 hours; however, in the presence of 3MA and 2mix, a significant induction of LMP was already evident after 12 hours. Moreover, the 2mix-induced LMP was further significantly increased upon inhibition of autophagy by 3MA after both 12 hours and 24 hours (Fig. 3C). Furthermore, 2mix-induced LMP was reversed by rapamycin by up to 46%. The 2mix-mediated LMP was confirmed by cathepsin D immunocytochemistry (Fig. 3D). Lysosomal cathepsin D granules were evident in untreated control cells, while most of the 2mix-treated cells exhibited a cytosolic diffuse pattern and only few cathepsin-positive granules remained in the lysosomes, indicating LMP mediated by 2mix (Fig. 3D). LMP was further examined by AO relocation test, in which cells were first stained with AO and then exposed to different treatments. As shown in Figure 3E, control cells showed bright lysosomal red and low cytosolic/nuclear AO green fluorescence, indicating intact lysosomal membranes, while cells treated with 2mix showed bright AO green fluorescence and reduced lysosomal red fluorescence, indicating LMP and relocation of AO from lysosomes to cytosol. Rapamycin pretreatment partially preserved lysosomal red fluorescence and decreased cytosolic relocation of AO induced by 2mix, while 3MA enhanced 2mix-induced LMP. As positive control, LMP was induced by CQ, which was further enhanced by 2mix (Fig. 3E). These results indicate that the cytoprotective effect persuaded by autophagy induction in the cell model is via the reduction of ROS and LMP.
Discussion
Apoptosis and autophagic cell death may play pivotal roles in plaque rupture and thrombosis of atherosclerotic lesions. However, the relationship and molecular interplay between autophagy and apoptosis in foam cells have not yet been investigated. In the present study, we demonstrate that autophagy dysfunction is involved in 7-oxysterol-induced apoptosis. Induction of autophagy by rapamycin protects the cells against apoptosis induced by 7-oxysterols by preventing LMP and reducing ROS levels. Moreover, autophagy induction reduces cellular lipid accumulation.
Lipid accumulation and apoptotic cell death are of importance in necrotic core formation and progression of atherosclerotic plaques. Oxysterols are cholesterol oxidation products that are present in human tissue and fluids, including plasma, lipoproteins, and coronary and carotid atherosclerotic lesions. 16 7-Oxysterols are the major cytotoxic components found in oxidized low-density lipoprotein and they can induce apoptosis of vascular wall cells, including monocytes, macrophages, and endothelial cells.2,3,14 Here, we found that 7-oxysterols induce autophagy dysfunction and the simultaneous occurrence of apoptotic and post-apoptotic necrotic cell death. Such dysfunctional autophagy may lead to further intracellular lipid accumulation.
The results from the present study suggest that 7-oxysterols induce both autophagy synthesis, as indicated by increased Atg5 and LC3 levels, and decreased autophagy degradation, as marked by the induction of p62. We consider that the earlier induced Atg5 and LC3 may have a cytoprotective effect; however, it is insufficient in the process. Additional induction of autophagy by rapamycin partially protected cells from 7-oxysterol-induced cell death, which was achieved by suppression of ROS and LMP. Rapamycin, a lipophilic and macrolide antibiotic, induces autophagy by inactivating the mammalian target of rapamycin (mTOR). 17 Rapamycin has been shown to counteract cellular ROS induced by mTOR hyperactivity, and removal of ROS reduced hyper-activated mTOR levels in hematopoietic stem cells. 18 Moreover, oral administration of rapamycin decreased plaque vulnerability in a rabbit atherosclerotic model associated with reduction of monocyte chemoattractant protein-1 and matrix metal-loproteinases. 18 Our results may propose a new mechanistic function for rapamycin in the interference of apoptosis in atherosclerosis.
Our unpublished results show that autophagy activity in early human atheroma lesions may be a transient self-defense mechanism, which declines following prolonged lipid oxidation and oxidative stress. It is not yet fully understood if autophagy activity plays a crucial role in maintaining atheroma plaque stability via regulation of lipid accumulation and necrotic core formation. In mice, lack of Atg5 augmented lipoprotein cholesterol ester retention due to decreased cholesterol efflux. 11 Our results revealed that rapamycin remarkably decreased cellular lipid accumulation in a 7-oxysterol-mediated cell death model. The mechanisms underlying such reduction of cellular lipids need further investigation.
In conclusion, 7-oxysterols induce not only apoptosis/post-apoptotic necrosis but also the formation of autophagic vacuoles, and the latter phenomenon may possibly be dysfunctional. Autophagy induction by rapamycin minimized 7-oxysterol-induced dysfunctional autophagy and cell death via the reduction of ROS, LMP, and cellular lipids. Our findings raise the possibility that autophagy may serve a protective role in the regulation of oxidized lipid-mediated cytotoxicity to limit necrotic core formation in atheroma progression.
Footnotes
Author Contributions
Conceived and designed the experiments: XMY and WL. Analyzed the data: WL, NSiraj and NSultana. Wrote the first draft of the manuscript: WL and XMY. Contributed to the writing of the manuscript: NSiraj, NSultana, LJW, and BG. Agree with manuscript results and conclusions: All authors. Made critical revisions and approved final version: WL and XMY. All authors reviewed and approved of the final manuscript.