
Abstract
The natural breakdown of cells, tissues, and organ systems is a significant consequence of aging and is at least partially caused by a decreased ability to tolerate environmental stressors. Based on quantitative ultrastructural analysis using transmission electron microscopy and computer imaging, we show significant differences in hepatocyte morphology between young and old rats during a 48-hr recovery period following a 2-day heat stress protocol. Mitochondrial injury was greater overall in old compared with young rats. Autophagy was observed in both young and old rats, with autophagy greater overall in old compared with young hepatocytes. Lipid peroxidation and protein nitration were evaluated by localization and quantification of 4-hydroxy-2-nonenal (4-HNE)–modified protein adducts and 3-nitrotyrosine (3-NT) levels, respectively. Levels of 3-NT but not 4-HNE-protein adducts were significantly elevated in hepatocytes of old rats in comparison with young at 90 min after heat stress, suggesting a major role for reactive nitrogen species in the pathology observed at this time point. These results show a differential response of hepatocyte mitochondria to heat stress with aging, as well as greater levels of both autophagic and nitrative damage in old vs young hepatocytes. This manuscript contains online supplemental material at http://www.jhc.org. Please visit this article online to view these materials.
T
ROS and RNS are both physiological messengers and normal byproducts of cellular metabolism. Under physiological conditions, the potentially injurious effects of ROS are kept in check by antioxidant enzymes (AEs) such as manganese superoxide dismutase (MnSOD), copper, zinc superoxide dismutase (CuZnSOD), catalase (CAT), and glutathione peroxidase (GPx). Several studies have suggested that, as an organism ages, ROS/RNS naturally begin to accumulate within cells, with ROS/RNS levels exceeding capacities of detoxifying AEs within cells. The resultant imbalance between ROS/RNS and AEs can cause increases in oxidative/nitrative stress within an organism; if the AE system is sufficiently overwhelmed, the increase in ROS/RNS and accumulation of oxidized macromolecules such as proteins, lipids, and nucleic acids cause damage to cells (Ames et al. 1993; Berlett and Stadtman 1997; Bejma and Ji 1999).
There is extensive experimental evidence that damage (especially related to oxidative stress) to proteins, DNA, membrane lipids, and cell organelles plays an important role in the aging process. Accumulation of damaged extracellular, intracellular, and membrane proteins may account for the age-associated malfunctioning of many biological processes and has been frequently used as a biomarker for aging (Stadtman 2001; Kregel and Zhang 2007). In addition, stress-induced cellular injury and death seem to be exaggerated with aging, and previous studies suggest that an age-related increase in oxidative stress contributes to these deleterious responses (reviewed by Finkel and Holbrook 2000). The goal of this study was to localize and quantify damage in rat hepatocytes at the subcellular level following a heat stress protocol and examine what role, if any, ROS/RNS play in the previously reported hepatocyte damage that occurs following heat stress (Hall et al. 2000). We chose to examine hepatocellular injury using biochemistry and electron microscopy (EM) techniques. Immunogold EM was used to localize and quantitate levels of 4-hydroxy-2-nonenal (4-HNE)–modified protein adducts and 3-nitrotyrosine (3-NT) using highly specific antibodies. Both 4-HNE (Toyokuni et al. 1995) and 3-NT (Haddad et al. 1994) have been shown to be highly specific markers of lipid peroxidation with subsequent protein adduction and protein nitration, respectively.
The mechanism of cell death that accompanies heat stress has not been conclusively determined to date. This study documents mitochondria and peroxisomes as prime targets for heat stress–induced hepatocyte injury and shows that mitochondria, but not peroxisomes, are more prone to injury in old vs young rats. Increased autophagy observed in old rats following heat stress is a new finding and may represent a response of the cell to remove lipid peroxidation protein adducts. Increased protein nitration was also observed in old rats in comparison with young rats following heat stress, and the possible relevance of this novel finding is discussed.
Summary of heat stress protocol for young (6 months old) and old (24 months old) rats

a Rats kept at ambient temperature (22–24C) but fitted with a colonic temperature probe.
b Zero-hr time point represents rats euthanized immediately after the second heat stress.
Tco, colonic temperature.
Materials and Methods
Animals and Heat Stress Protocol
Young (6 months old) and old (24 months old) male Fischer 344 rats (National Institute on Aging; Bethesda, MD) were used in these experiments. Rats were housed in The University of Iowa Animal Care Facility, and all experimental procedures were approved by the Institutional Animal Care and Use Committee. Animals were maintained at 22–24C on a 12:12-hr light-dark cycle and provided food (standard rat chow) and water ad libitum. Before experimentation, rats were randomly placed into various experimental groups (n=3 rats/group).
All rats were handled daily and familiarized with a colonic temperature (Tco) probe during the week before the heat-stress protocol. On the day of the first heat exposure, each rat was fitted with a thermistor temperature probe inserted 6–7 cm into the colon and then was placed in a plastic cage (45 × 25 × 20 cm), conscious and unrestrained. Tco was continuously monitored on a digital display. A baseline Tco (37.0–38.0C for both age groups) was established over a 30-min control period for each rat, followed by a heating protocol that has been previously described (Hall et al. 2000; Zhang et al. 2003). A summary of the heating protocol is shown in Table 1. Sham-heated control rats were handled identically to experimental rats, with the exception that ambient temperature was maintained at 22–24C. At 0 hr (i.e., immediately after the second heating), 30 min, 60 min, 90 min, 6 hr, 24 hr, and 48 hr after the second heating, rats were administered an overdose of pentobarbital sodium (80 mg/kg, IP). Livers were collected and immediately frozen for biochemical analyses or placed in appropriate fixatives for routine or immunogold EM.
Routine EM
Liver tissues were collected, cut into 1-mm3 pieces, immediately immersed in 2.5% glutaraldehyde in phosphate buffer (pH 7.2), and fixed overnight at 4C. Tissues were rinsed in Sorenson's phosphate wash buffer, postfixed in Caulfield's osmium tetroxide plus sucrose, and rinsed again in wash buffer. All supplies and reagents, unless otherwise noted, were from Electron Microscopy Sciences (Hatfield, PA). Tissues were dehydrated in a graded series of ethanols and propylene oxide and embedded in BEEM capsules using EMbed 812 resin. Polymerization was thermally induced overnight in a 60C oven. Thin sections (70–80 nm) were cut and mounted on copper grids, stained with filtered 7.7% uranyl acetate, and counterstained with Reynolds lead citrate. Tissue sections were observed using a transmission electron microscope (H-600; Hitachi, Schaumburg, IL) operated at 75 kV. Three young and three old rats (n=3) were analyzed for each time point with two exceptions (see below). Micrographs of 20 hepatocytes from each rat (total of 60 per time point) were taken randomly at low magnification (×3000) by scanning the tissue grid and taking one micrograph per grid square; areas containing any sampling artifact (i.e., “dirt”, staining precipitates, or tissue folds) were avoided if at all possible. The liver specimen from one of the old 0-hr rats and one of the old 30-min rats exhibited uniformly poor morphology, most likely an artifact of insufficient penetration of the glutaraldehyde fixative. These rats were excluded from the analysis to avoid any false bias of the results; therefore, instead of 20 hepatocytes from three rats (total of 60 hepatocytes per time point), 30 hepatocytes from the remaining two rats at both 0 hr and 30 min were analyzed to keep the total number of hepatocytes studied at each time point the same throughout the study.
Morphometric Quantitation of Hepatocytes Using Transmission EM
To analyze hepatocytes for either damage or subcellular localization of 4-HNE and 3-NT, electron micrographs were developed and converted to digital images using a Perfection 4870 (Epson; Long Beach, CA) high-resolution negative scanner in 8-bit grayscale with the unsharp mask filter option selected. Digital images of hepatocytes were observed on a PC computer (OptiPlex GX280; Dell, Round Rock, TX) using Photoshop Elements 2.0 (Adobe Systems; San Jose, CA) to manually count gold beads and identify subcellular areas of interest and/or injury. The image analysis program Scion Image Beta 4.02 (Scion Corporation; Frederick, MD) was used to measure the relative area (μm2) of the hepatocytes and subcellular organelles. An analysis was performed to determine the difference (if any) in the occurrence of autophagy and damage to mitochondria and peroxisomes in young vs old rats across the entire recovery time course (sum of control through 48 hr after heat stress). The average percent abnormal area of 480 cells (8 time points × 3 rats per time point × 20 hepatocytes analyzed per rat) for both young and old was calculated, and data are presented in Results.
Criteria for Analysis of Damage to Hepatocytes
There were several subcellular compartments analyzed for damage, including nuclei and nucleoli, mitochondria, peroxisomes, myelin figures, autophagosomes, and cytoplasm. Well-established criteria for ultrastructural morphology and damage have been documented (Ghadially 1997) and were used to define damage in each subcellular compartment measured or observed in this study (see online Supplemental Table ST1). Organelles observed in the cytoplasm that were not measured quantitatively for damage include smooth and rough endoplasmic reticulum, glycogen, Golgi complexes, vacuoles of undetermined origin, cytoplasmic granules, and membrane vesicles; these structures are included with the cytoplasm in the area calculated as cytoplasmic area. Any significant aberrations in the morphology of these latter cytoplasmic features are noted in Results.
Immunogold EM
Liver tissues were collected, cut into 1-mm3 pieces, immersed in Carson Millonig's fixative (4% formaldehyde in 0.16 M monobasic sodium phosphate buffer, pH 7.2) for 1 hr at room temperature, and processed for immunogold EM as previously described in detail (Oberley 2002). Briefly, samples were dehydrated in graded ethanols and embedded in LR White resin (Electron Microscopy Sciences) in sealed gelatin capsules. Polymerization was thermally induced overnight in a 55C oven. Ultrathin sections (70–80 nm) were cut and mounted on nickel grids for postembedding immunogold procedures. Sections were preincubated at room temperature with filtered TBS, pH 7.6, followed by filtered 0.5% acetylated BSA (BSA-C; Aurion, Wageningen, The Netherlands) in PBS, pH 7.4, to block nonspecific antibody binding sites. Sections were rinsed in filtered PBS at room temperature and incubated with primary antibody overnight at 4C in a humidified chamber. Primary antibodies [mouse anti-4-HNE protein adducts (JaICA; Shizuoka, Japan) and rabbit anti-3-NT (Upstate Biotechnology; Lake Placid, NY)] were diluted to optimal concentrations (1:140 4-HNE; 1:600 3-NT) with phosphate-buffered antibody diluent (ScyTek Laboratories; Logan, UT) as determined by titration analyses using suitable positive control tissues. Normal mouse or rabbit serums (Dako; Carpinteria, CA) were used in place of primary antibodies as negative controls. After overnight antibody incubation, sections were allowed to warm at room temperature, washed with filtered TBS wash buffer, and incubated with either anti-mouse IgG (1:50) or anti-rabbit IgG (1:75) 15-nm gold-conjugated secondary antibody (B.B. International; Cardiff, UK). The secondary antibodies were newly purchased and centrifuged immediately before use to minimize the occurrence of gold aggregates. All samples for both 3-NT and 4-HNE were immunolabeled at the same time to keep conditions constant. Sections were washed in filtered TBS wash buffer followed by filtered distilled water.
To avoid false-positive label of the 3-NT antibody because of reduction-oxidation (redox) reactions initiated in tissues by atmospheric oxygen/nitrogen, tissue sections were placed under vacuum immediately after sectioning; all preincubation steps with TBS, BSA-C, PBS, and overnight incubation with primary antibody were performed under vacuum. Sections were counter-stained with filtered 7.7% aqueous uranyl acetate and examined with a transmission electron microscope (H-600; Hitachi) operated at 75 kV. Young and old control and 90-min post–heat stress rats (n=3 per group) were analyzed. Using low magnifications so that gold beads were not discernible, 10 hepatocytes were randomly selected for each time point and photographed. Areas containing any sampling artifact were avoided if at all possible. Several previous studies have used these immunogold techniques for quantification of specific proteins in subcellular compartments (Chaiswing et al. 2004; Lucocq et al. 2004; Mayhew et al. 2004; Nithipongvanitch et al. 2007).
ATP Biochemistry
ATP extraction was achieved by perchloric acid digestion. An exact mass of finely powdered frozen (non-lyophilized) liver tissue (ranging from 0.05 to 0.10 g) was added to 285 μl of 15% perchloric acid. Samples were incubated with gentle agitation for 20 min at 4C. Cellular debris was removed by centrifugation at 2000 × g for 15 min at 4C. The supernatant was brought to a pH of 7.8 by addition of 1 M dibasic potassium phosphate solution. Samples were quantitatively collected by rinsing the pH probe with 2 ml of 15 mM Tris-HCl buffer (pH 7.8). Precipitate was removed by centrifugation at 2000 × g for 20 min at 4C. Supernatants were carefully collected and volumes were measured. ATP concentration in the extracts was measured using the ATP bioluminescent assay kit (Sigma-Aldrich; St. Louis, MO). Samples were diluted 1000-fold and combined in a 1:1 ratio with the ATP reaction mix. Luminescence was immediately measured and integrated over 6 sec using a Luminoskan Ascent (Thermo Scientific; Waltham, MA) luminometer. Light was reported in relative light units and compared against a standard curve to determine total ATP concentration in the liver tissue. ATP levels were analyzed in young vs old rats across the entire recovery time course (sum of control through 48 hr after heat stress). The average nanomoles of ATP per milligram of tissue collected from at least three separate measurements per time point for both young and old rats was calculated, and data are presented in Results.
Statistical Analysis
Data were analyzed using the independent Student's t-test and one-way ANOVA with the least significant difference post hoc test for multiple comparisons using SPSS for Windows 14.0 (SPSS; Chicago, IL). For immunogold data, ratios between control and 90-min time points were compared with non-parametric bootstrap analysis using R 2.3.1 (The R Foundation for Statistical Computing, www.r-project.org). SEs for ratios were calculated using propagation of error theory. Statistical data are represented graphically with all results. Mean values are represented as ±SEM and were considered significant at p<0.05.
Results
Ultrastructural Analysis of Rat Hepatocytes Following Heat Stress
Both young and old control (sham-heated) rats displayed insignificant injury to subcellular organelles, membranes, or cytoplasm. A typical hepatocyte from a control young rat is illustrated in online Supplemental Figure SF1. At most time points, the total relative hepatocyte area (μm2) from old rats was on average 10–20% less than that of young rats, and both young and old rats throughout the 48-hr recovery period following heat stress had relative hepatocellular areas ∼10–12% less than their age-matched controls (data not shown). These morphological changes are likely caused by a combination of atrophy in the older rat hepatocytes and direct or indirect hyperthermically induced pathological changes.
Within the first 90 min of recovery following heat stress, hepatocytes from both young and old rats began to exhibit signs of pathological injury and autophagic degradation. At 0 hr (rats euthanized immediately following the second heat stress), hepatocytes from both young and old rats showed a slight increase in overall damage to organelles and mild cytoplasmic vacuolization, along with an apparent decrease in glycogen and disorganization of the rough endoplasmic reticulum. One striking feature was injury to intracellular membranes as indicated by the presence of myelin figures in the cell cytoplasm, along organelle membranes, and on the peripheries of lipid droplets. A representative example of myelin figures is shown in Figure 1A, found in an old rat at 0 hr. Young and old rats throughout the recovery time course had an average 2- to 3-fold increase in relative area of myelin figures compared with untreated young cohorts (data not shown); however, when averaged across the sum of the time course, there was no significant difference in relative area of myelin figures between young and old rats (data not shown). This implies the observed increase in myelin figures was a pathological change caused by heat stress, which was not modulated by age.
Ultrastructural changes observed at 30 min after heat stress involved noticeable injury to several subcellular regions in both young and old rats. Mitochondria showed progressive loss of cristae, swelling, and myelin figures (Figure 1B), with irreversible mitochondrial injury first observed at 90 min after heat stress; such injury was indicated by the presence of flocculent (“woolly”) densities (Figure 1C). Damage to peroxisomes featured gaps in the unit membrane and a decrease in the intensity (“ghosting”) of the granular matrix (Figure 1D). There was an approximate 2-fold increase in ultrastructural autophagy visible at 30 min, as evidenced primarily by a significant increase in the number of autophagic vacuoles present in the cell cytoplasm at 30 min (Figure 2A) and by the presence of autophagolysosomes (Figure 2B) and a small amount of residual bodies and lipofuscin (Figure 2C) at later time points; Figure 2B is from an old rat at 60 min and Figure 2C is from an old rat at 24 hr after heat stress. Interestingly, the presence of an autophagy protein [rat microtubule-associated protein 1 light chain 3 (LC3)] known to directly correlate with the extent of autophagosome formation (Kabeya et al. 2000) was detected by Western blot immediately (0 h) following the second heat stress (see online Supplemental Figure SF2 and Supplementary Methods). Both the cytosolic form (LC3-I) and the membrane-bound form (LC3-II) of LC3 were significantly increased in old rats immediately following heat stress, suggesting there were biochemical alterations in autophagy occurring before visible evidence of ultrastructural changes was observed at 30 min. Visible swelling and myelin figure formation in the Golgi complexes and smooth endoplasmic reticulum were observed, along with an apparent decrease in the area and organization of rough endoplasmic reticulum and glycogen.
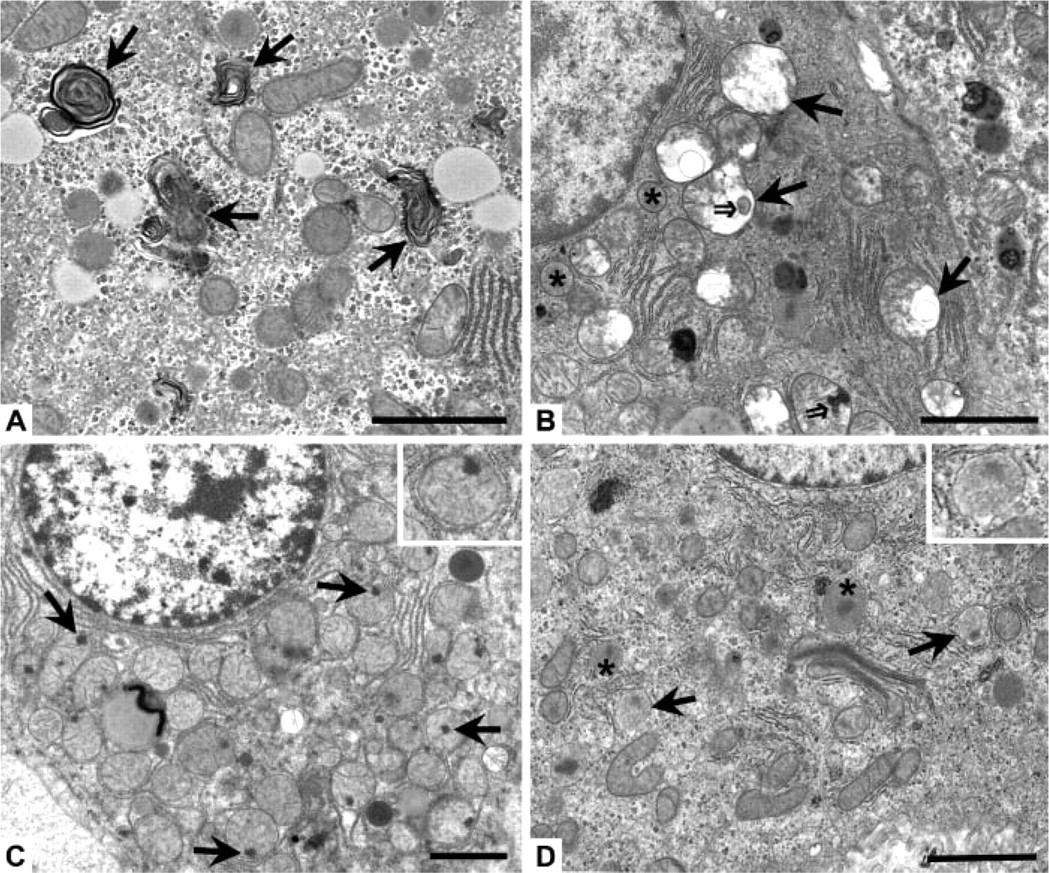
Representative micrographs of subcellular hepatocyte morphology during recovery from heat stress. (
A significant quantitative increase in mitochondrial damage was noted in both young and old rats throughout the first 6 hr of recovery from heat stress (Figure 3A), with mitochondrial damage increasing in both frequency and severity over this time period. Mitochondrial damage peaked in old rats (90 min) slightly later than young rats, who reached peak mitochondrial damage at 60 min after heat stress. In addition, the extent of mitochondrial injury in old rats was greater by ∼25% throughout the sum of the recovery time course compared with young cohorts (Figure 3B). The amount of autophagy increased 2-fold in both young and old rats at 30 min, with old rats showing a dramatic increase in autophagy at 60 min (Figure 3C). Similar to mitochondrial injury, the overall amount of autophagy in old rats was ∼25% higher than in young rats (Figure 3D) throughout the recovery time course. There were no significant differences between young and old rats in the amount of damage to peroxisomes at any time point during recovery from heat stress (Figure 3E); however, there was a significant decline in peroxisomal damage in young rats from 60 min through 6 hr compared with age-matched controls. There was no significant difference in peroxisomal damage in old vs young rats when compared across the sum of the time course (Figure 3F).
At 60–90 min after heat stress, extensive subcellular injury and focal signs of inflammation were observed. Progressive clumping of glycogen and swelling of smooth endoplasmic reticulum was noted. Resident macrophages (Kupffer cells) containing degenerating cellular material and cholesterol crystals were identified in areas adjacent to hepatocytes. At 6 hr after heat stress, vacuolization and membrane injury (very large myelin figures) were identified for the first time at the cell surface of hepatocytes, most often at the bile canaliculi. Apoptotic hepatocytes were noted occasionally in old rats at 60–90 min, as well as numerous red blood cells in the interstitial spaces between hepatocytes. Mitochondria from old rats appeared to have less edema and more abundant cristae at 6 hr than noted at 90 min of recovery, yet some mitochondria still contained the flocculent densities observed previously. The presence of both glycogen and lipid with normal morphological characteristics were identified in many hepatocytes from both young and old rats.
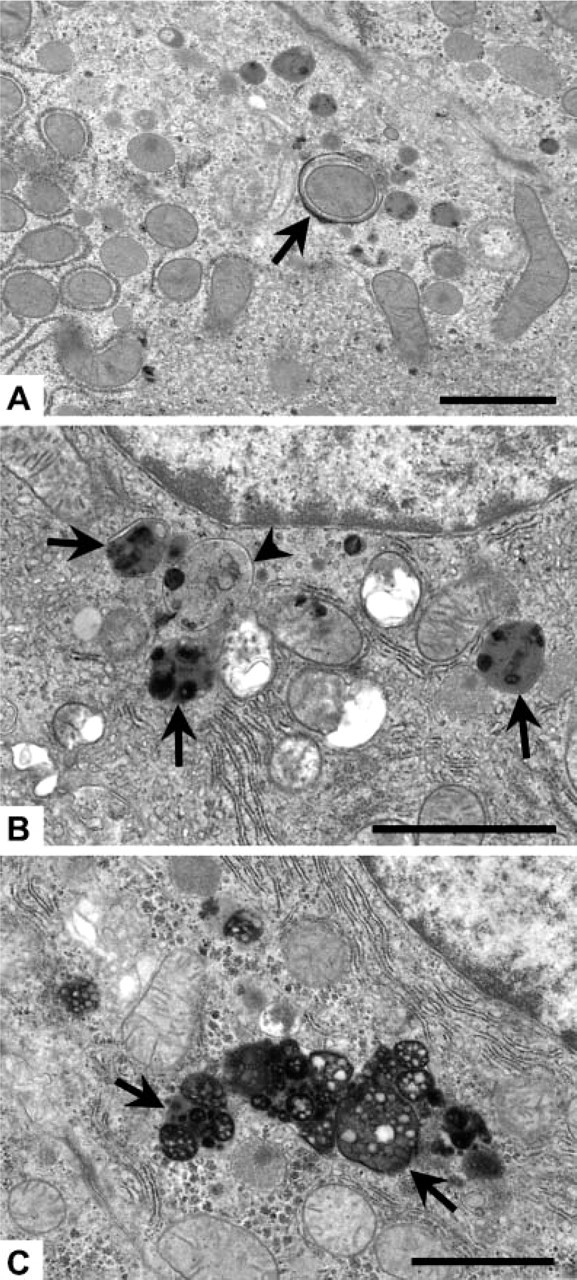
Representative micrographs of three stages of autophagy identified in hepatocytes and used for quantification. (
At 24 hr after heat stress, numerous hepatocytes from old rats were observed to have less damaged morphology than noted from 90 min to 6 hr, evidenced by an increased number of mitochondria with little to no swelling and more abundant cristae, along with an increase in glycogen and rough endoplasmic reticulum profiles. Focal areas of tissue necrosis were observed in liver from both young and old rats at this time point. At 48 hr after heat stress, hepatocytes from both young and old rats had morphologies similar to those of hepatocytes noted at 24 hr. The rough endoplasmic reticulum of several hepatocytes from young rats, however, appeared to be disintegrating and lacked lamellar cisternae, leaving behind clumps of ribosomes in the cytoplasm. Dead cells were infrequently observed in both young and old rats, presumably because of removal by blood inflammatory or parenchymal Kupffer cells.
Analysis of Oxidative and Nitrative Damage Using Immunogold EM
The most intense subcellular injury was identified at 90 min after heat stress; therefore, our immunogold studies were focused on this time point. Antibodies against 4-HNE–modified proteins and 3-NT were used to determine whether the damage observed by light and routine EM was oxidative and/or oxidative plus nitrative, respectively. Young and old rat livers labeled with antibody to 4-HNE protein adducts (Figures 4A–4D) showed the highest labeling density in mitochondria and peroxisomes—approximately twice the amount of this lipid peroxidation–protein adduct found in the cytoplasm and nuclei. Localization of 3-NT in young and old rats was also higher overall in mitochondria and peroxisomes, with slightly lower label in the cytoplasm and nuclei (Figures 4E–4H). Quantitation of oxidative/nitrative damage showed overall higher levels of 4-HNE protein adducts in many subcellular compartments of old rats in comparison to their young cohorts, although the difference was only significant (p<0.05) in the peroxisomes (Figure 5A). When ratios of 90-min young and old were calculated against controls, however, there were no significant changes or differences in 4-HNE protein adducts between young and old rats in any subcellular compartment (Figure 5B). Levels of 3-NT were lower in old control rats compared with young controls, whereas old rats at 90 min had significantly higher levels than young cohorts (Figure 5C). Ratios of 90 min young and old compared with age-matched controls showed a significant 2-fold increase in 3-NT in all subcellular compartments of old rats compared with young cohorts (Figure 5D).
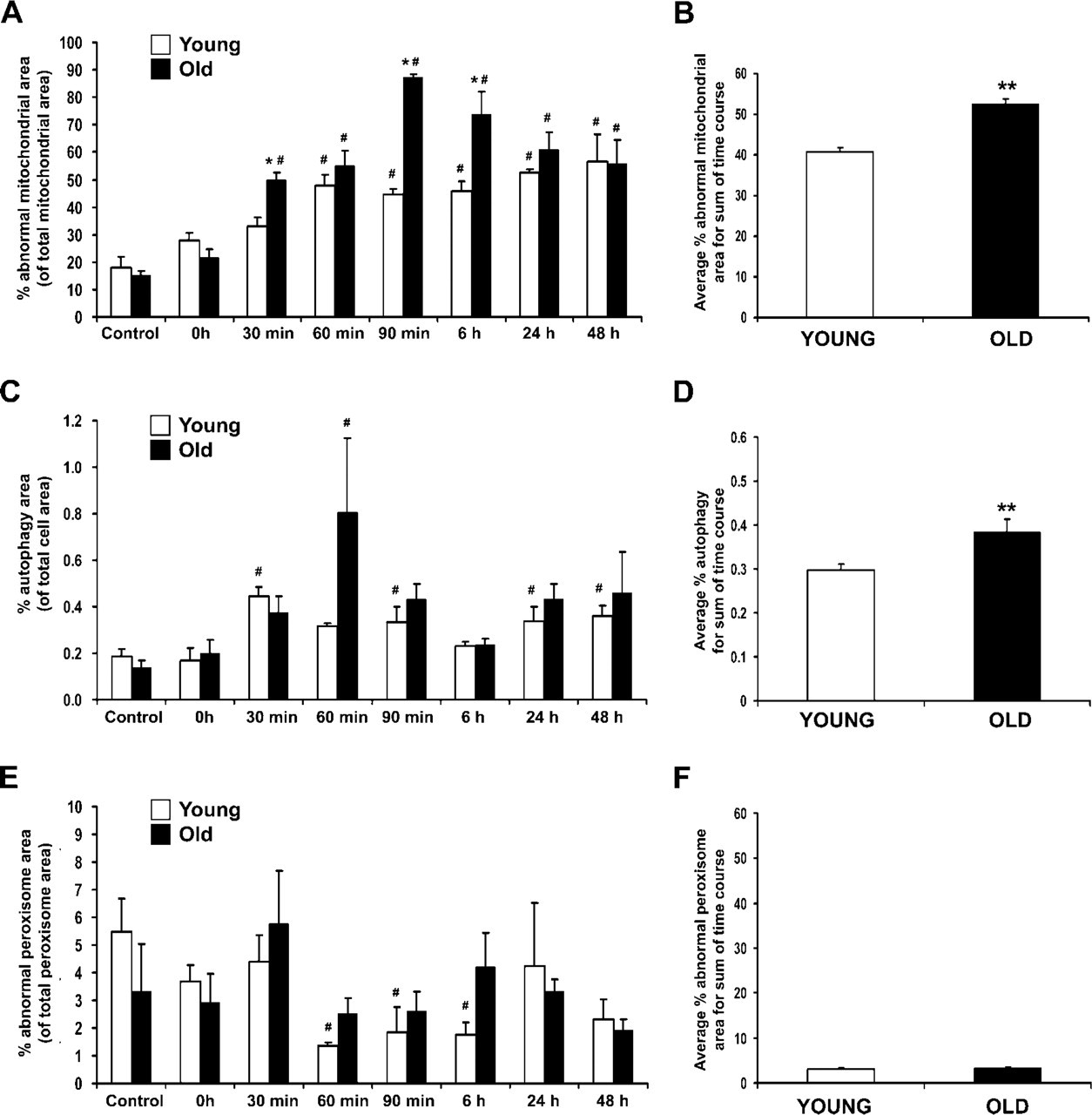
Quantitative analysis of subcellular hepatocyte damage following heat stress. (
ATP Biochemistry
To measure the effect of heat on the energy metabolism of young and old hepatocytes, an assay using the ATP-dependent enzyme firefly luciferase was employed. Throughout much of the recovery time course in this study, there was a significant decrease in the amount of ATP in liver tissues from old rats compared with age-matched controls and/or young cohorts (Figure 6A). The amount of ATP in liver samples from young rats increased almost 2-fold at the 12- to 48-hr time points, whereas tissues from old rats increased almost 3-fold at 24 hr following heat stress. When analyzed across the sum of the time course, old rats averaged one third less ATP than their young cohorts (Figure 6B).
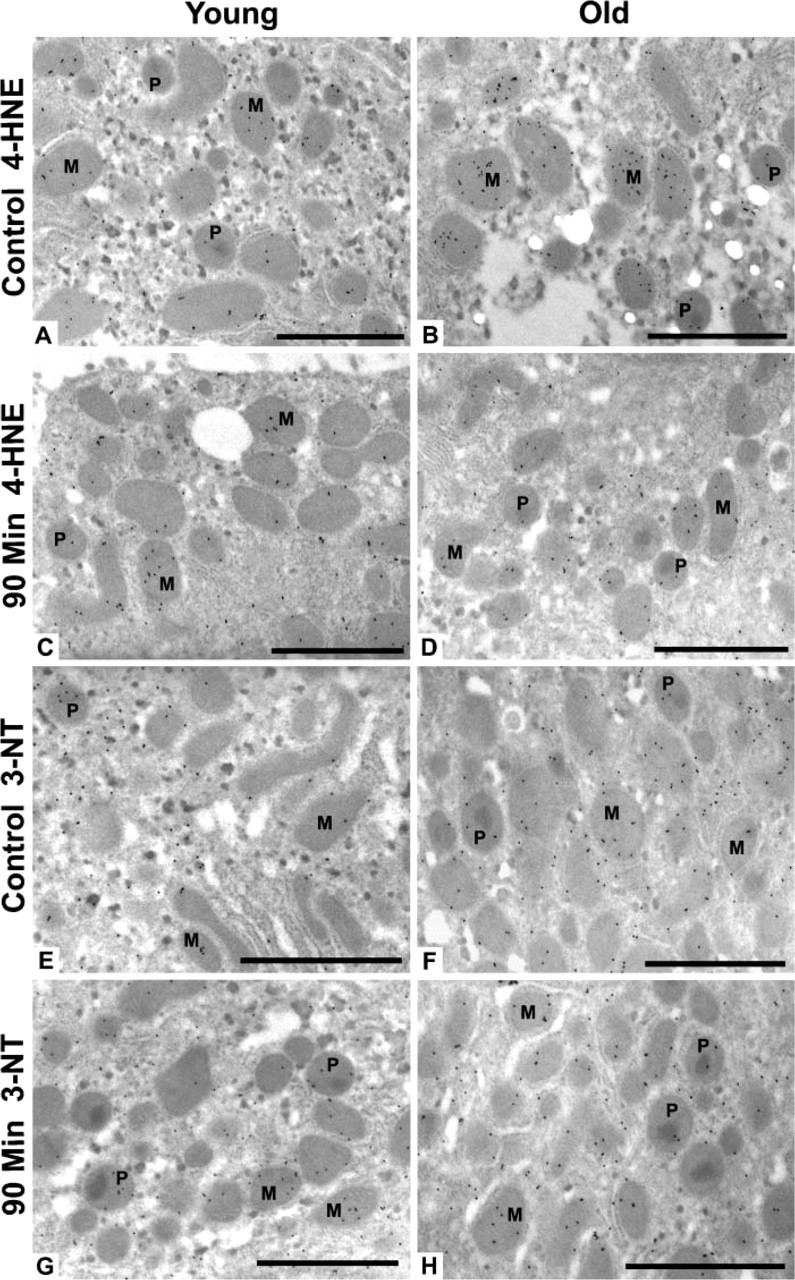
Representative micrographs of 4-hydroxy-2-nonenal (4-HNE) protein adducts and 3-nitrotyrosine (3-NT) gold labeling in rat hepatocytes. Control young (
Discussion
This study showed prominent morphological and subcellular injury in both young and old rat hepatocytes following heat stress. The extent of damage to hepatic ultrastructure and the subcellular localization of oxidative/nitrative damage products were found to be quite different depending on age following a 2-day heat stress protocol. The impaired response to heat stress observed in old rats could be caused by a failure to induce heat shock proteins (HSPs) such as HSP70 and HSP32, or other stress signaling pathways (Fargnoli et al. 1990; Nitta et al. 1994; Heydari et al. 2000; Zhang et al. 2003; Tandara et al. 2006). Because HSP70 is believed to promote the correct refolding of denatured or unfolded proteins damaged by stress and can also act as a chaperone in the ubiquitin–proteasome degradation pathway (Riezman 2004), the failure of senescent organisms to properly induce the expression of HSP70 and other HSPs would lead to the accumulation of damaged proteins that may be toxic to the cell (Kregel 2002; Riezman 2004). Such lipid–protein aggregates (residual bodies and lipofuscin) were observed in this study.
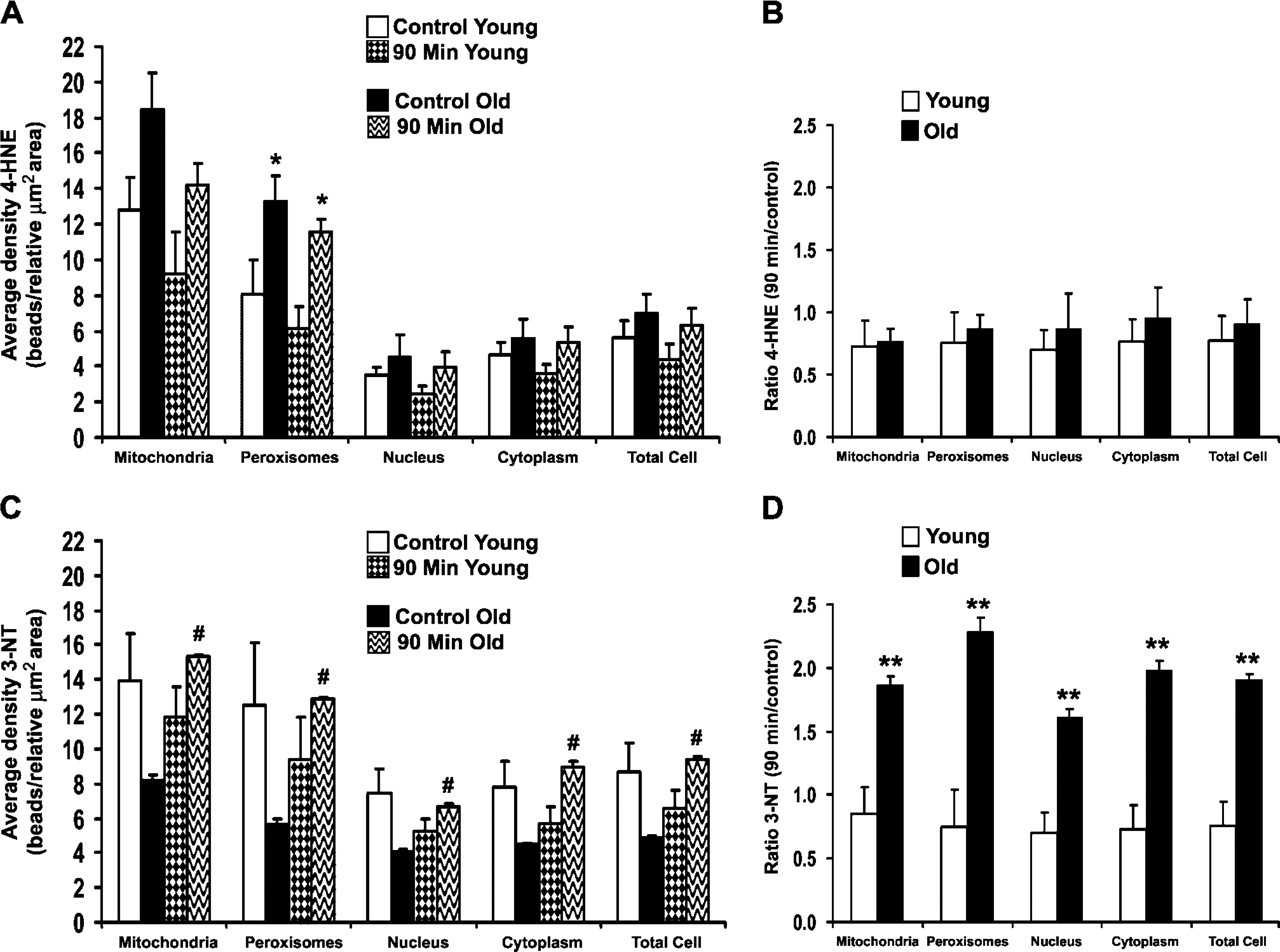
Quantitative analysis of 4-hydroxy-2-nonenal (4-HNE) protein adducts and 3-nitrotyrosine (3-NT) gold labeling in rat hepatocytes. (
It is well established that lipofuscin is found to accumulate in cells of older organisms, particularly in terminally differentiated tissues such as heart and neurons (Terman and Brunk 2004; Cuervo et al. 2005; Terman and Brunk 2006; Terman et al. 2006a, b). Lipofuscin represents a collection of oxidatively modified proteins and degraded lipid material that cannot be further degraded by lysosomal hydrolases or be exocytosed. However, some iron-containing metallo-proteins degrade, and the resulting release of low molecular mass iron will cause Fenton-type chemistry in the presence of hydrogen peroxide. The iron-catalyzed reaction of hydrogen peroxide with superoxide radical (O2•-) produces the extremely toxic and reactive hydroxyl radical (•HO); this produces aldehydes [e.g., malondialdehyde (MDA)] and finally results in lipofuscin (Terman and Brunk 2004). A previous study in our laboratory showed that lipofuscin in the prostate contained 4-HNE protein adducts (Oberley et al. 2000). In vivo, severe ATP depletion can cause destabilization and aggregation of many proteins (Kabakov et al. 2002). If HSP70, a known ATP-dependent chaperone induced by the appearance of denatured proteins within a cell following stress, is not being properly induced in old rats following heat stress as previous studies have shown (Fargnoli et al. 1990; Nitta et al. 1994; Hall et al. 2000; Heydari et al. 2000; Zhang et al. 2006), the resultant subcellular stress caused by the toxic accumulation of protein aggregates could be significant enough to cause damage to mitochondria, peroxisomes, rough endoplasmic reticulum, and membrane lipids similar to that observed in this study.
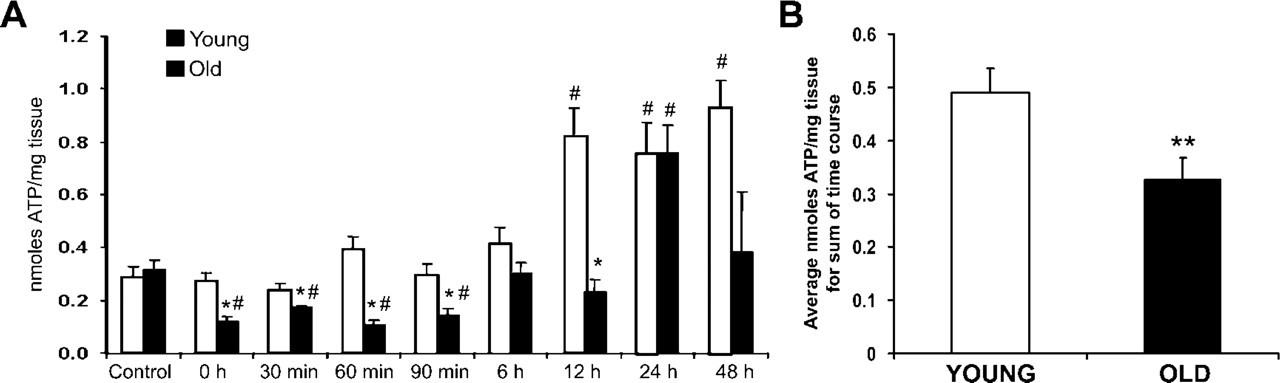
ATP biochemistry. (
Decreased ATP levels in old rats compared with young throughout most of the recovery time course indicated an overall decreased ability of senescent animals to compensate for a loss in energy metabolism, correlating with increased mitochondrial damage observed following heat stress. However, there were significant increases in ATP levels at 12 (young) and 24 hr (young and old), and these were time points in recovery at which mitochondrial damage appeared to decline in old rats; this could imply an improvement toward normal cellular metabolism, perhaps caused by significant upregulation of autophagic protein degradation measured at earlier time points of recovery and subsequent mitochondrial repair or renewal. Kroemer and Jaattela (2005) suggest that the mere presence of autophagosomes within cells is not proof that increased autophagy has occurred; however, we directly quantitated the percent cell area occupied by the sum of three separate stages of autophagy that were clearly identifiable within the hepatocytes. Taken together with the significant increases in the autophagosome-associated LC3 protein observed at early recovery time points, we feel confident that the amount of autophagy was indeed significantly increased following heat stress.
Also supporting the theory of decreased tolerance to hyperthermia with age was a key pathological feature observed in the mitochondria of old rats at 90 min through 6 hr of recovery: round electrondense inclusions located near the inner mitochondrial membrane with morphology that closely resembled that of flocculent (“woolly”) densities. Development of a significant number of flocculent densities is thought to be one of the most reliable early manifestations of irreversible cell injury, cell death, and ensuing necrosis (Ghadially 1997). Quantitative analysis confirmed that mitochondrial damage in old rats was greatest from 60 min through 6 hr following heat stress, a time period that directly correlates with the appearance of flocculent densities in the mitochondria of senescent rats. These results suggest that not only is pathology increased in older hepatocytes, but at least some of the damage is irreversible.
Immunogold EM was used to localize specific antibodies to 4-HNE protein adducts and 3-NT for analysis of lipid peroxidation protein adducts and protein nitration, respectively. 4-HNE is a major oxidative damage product that is the end result of the breakdown of polyunsaturated fatty acids and related esters (Benedetti et al. 1980). 4-HNE is also a cytotoxic aldehyde that reacts easily with the sulfhydryl groups of proteins and low molecular mass thiols such as glutathione (GSH). Spitz et al. (1991) have shown that GSH is a key player in the detoxification of intracellular 4-HNE, and 4-HNE also reacts rapidly with cysteine, lysine, and histidine protein residues. Such reactions have been shown to inactivate proteins and enzymes, inhibit synthesis of protein, DNA, and RNA, and cause damage to lipid biomolecules (Esterbauer et al. 1991). A previous study by our laboratory (Zhang et al. 2003) showed significant increases in both free (non–protein bound) MDA and 4-hydroxyalkenal (HAE), markers of lipid peroxidation, in old vs young rats following heat stress. Ratio and bootstrap analysis in this study, however, showed no significant differences between young and old rats in the amount of 4-HNE–modified proteins in any subcellular compartment after 90 min of recovery from heat stress. Because the specific antibody used in this study only recognizes 4-HNE after it has bound to protein and not free 4-HNE (Toyokuni et al. 1995), we were able to show that not only were free radicals being produced in hepatocytes following heat stress, but there was resultant oxidation of proteins in both young and old rats. There are several possible explanations for these divergent results. First, 4-HNE–modified proteins appear very early following oxidative injury, significantly preceding histopathological and clinical indicators of tissue injury (Oberley 2002). The time point examined (90 min) in this study could therefore be too late for optimal measurement of oxidative damage following heat stress. A second reason could be that HSPs induced following the first heat stress were “masking” proteins so that either the 4-HNE antibody could not recognize the damaged proteins or the HSPs physically blocked 4-HNE from binding to proteins. The most likely explanation for the presence of free but not protein-bound lipid peroxidation products is the activation of autophagy by oxidized lipid-protein adducts with attendant enzyme catalysis by degradative enzymes in lysosomes. Additional studies will be necessary to prove this hypothesis.
Similar to 4-HNE protein adducts, the majority of protein nitration in hepatocytes as measured by 3-NT was found to localize primarily in the mitochondria and peroxisomes of young and old rats. We found an approximate 2-fold increase in 3-NT levels of old rats compared with young cohorts in all subcellular compartments measured, implying some sort of imbalance had developed involving reactive nitrogen species. An increase in 3-NT could occur because of augmented RNS production or decreased denitration by enzymes as yet unidentified. An increase in peroxynitrite (ONOO-) levels may be caused by reaction of O2•- with nitric oxide (NO•) and subsequent adduction to protein tyrosine residues (Beckman and Koppenol 1996). There was also higher expression of 3-NT in young control rats compared with old cohorts, a phenomenon best explained by the role of NO• in numerous physiological processes within cells (Monteiro et al. 2008). NO is a lipophilic, highly diffusible free radical that, when generated locally within mitochondria, can reversibly inhibit cytochrome-c oxidase (associated with complex IV of the mitochondrial electron transport chain) and cause leakage of O2•-, a free radical normally scavenged by superoxide dismutase (SOD) (Richter et al. 1995; Beckman and Koppenol 1996). If NO• was produced in high enough concentrations equivalent to endogenous levels of SOD, mitochondria would be irreversibly injured because the O2•- would preferentially react with NO• rather than SOD to produce ONOO-, a highly reactive and potentially cytotoxic molecule (Ischiropoulos et al. 1992). The stability of ONOO- allows it to readily diffuse through a cell and interact with many biological targets, causing DNA condensation and damage, inhibition of protein synthesis, lipid and protein oxidation, and protein nitration (Rubbo et al. 1994; Kim et al. 1995). Results from this study indicate an approximate 2-fold increase in the amount of 3-NT found in old rats compared with young. When taking into account results from Leeuwenburgh et al. (1998) that showed no significant increase in physiological levels of 3-NT found in livers of old rats compared with young cohorts, results from this study would indicate the observed 2-fold increase in 3-NT in old rats was a consequence of heat stress and not a function of age.
Many studies have shown that NO• readily oxidizes low molecular mass thiols, in particular, GSH. Chen et al. (2003) hypothesized that the level of redox stress would be a primary determinant of NO•-mediated toxicity in hepatocytes and that mitochondrial targets, normally insensitive to NO•-mediated toxicity, would be targeted under conditions of redox stress. They found that depletion of GSH increased the level of oxidative stress caused by NO• and reduced ATP content, mitochondrial membrane potential, and mitochondrial respiratory enzyme activities. Results from our previous study (Zhang et al. 2003) indicated that the GSH/oxidized glutathione (GSSG) ratio was ∼60% greater in young control animals compared with their old cohorts, and old animals showed no significant changes in GSH/GSSG ratio in response to heat stress. With lower physiological levels of GSH present to buffer the effects of NO•, old rats would be more susceptible to oxidative injury and protein nitration than their young cohorts. Previous studies by Kim et al. (1995, 1997) found that low-level induction of NO• expression using S-nitroso-N-acetylpenicillamine (SNAP) protected cultured hepatocytes from oxidative injury and tumor necrosis factor α–induced apoptotic cell death by induction of HSP70 and HSP32. These results corroborate data in this study and previous findings from our laboratory (Hall et al. 2000; Zhang et al. 2006) that aged animals have higher levels of redox stress and are ill prepared to handle additional stresses brought on by factors such as hyperthermia. The first study by Hall et al. (2000) examined both the constitutive (HSC70) and inducible (HSP70) forms of HSP70 by Western blot and showed that the magnitude of the HSP70 response was significantly reduced with age during a 48-hr recovery from heat stress. Strong HSP70 expression was noted throughout the recovery time course in young rats; however, the initial elevation of HSP70 at 2 hr in old rats decreased significantly during later time points of recovery. The second paper by Zhang et al. (2006) measured HSP70 mRNA expression at the transcriptional level by semiquantitative RT-PCR analysis. Results from this experiment showed that both young and old rats are capable of upregulating HSP70 mRNA transcription to a similar level immediately following heat stress (0 h), whereas levels of HSP70 mRNA in old rats decreased significantly at 2 hr after heat stress.
The mechanisms underlying the transient and/or permanent pathological changes involved in the hepatocellular response to heat stress remain complex. In this study, we assessed damage caused by protein oxidation and nitration by using specific antibodies against 4-HNE protein adducts and 3-NT. The presence of increased amounts of 4-HNE–modified proteins and 3-NT, both highly reactive and potentially cytotoxic molecules, indicated a state of persistent oxidative and nitrative stress in hepatocytes of old rats following heat stress.
Footnotes
Acknowledgements
This work was supported in part by a grant from the National Institutes of Health (Grant AG012350 to KCK and TDO). The work was supported in part by resources and the use of facilities at the William S. Middleton Memorial Veterans Hospital, Madison WI.
We acknowledge the technical assistance of Joan Sempf, Ryan Kolarik, and Nathan Shaw.