
Abstract
Both apoptosis and autophagy are highly conserved processes that besides their role in the maintenance of the organismal and cellular homeostasis serve as a main target of tumor therapeutics. Although their important roles in the modulation of tumor therapeutic strategies have been widely reported, the molecular actions of both apoptosis and autophagy are counteracted by cancer protective mechanisms. While apoptosis is a tightly regulated process that is implicated in the removal of damaged or unwanted cells, autophagy is a cellular catabolic pathway that is involved in lysosomal degradation and recycling of proteins and organelles, and thereby is considered an important survival/protective mechanism for cancer cells in response to metabolic stress or chemotherapy. Although the relationship between autophagy and cell death is very complicated and has not been characterized in detail, the molecular mechanisms that control this relationship are considered to be a relevant target for the development of a therapeutic strategy for tumor treatment. In this review, we focus on the molecular mechanisms of apoptosis, autophagy, and those of the crosstalk between apoptosis and autophagy in order to provide insight into the molecular mechanisms that may be essential for the balance between cell survival and death as well as their role as targets for the development of novel therapeutic approaches.
Introduction
Both apoptosis and autophagy are highly conserved processes that in addition to their role in maintenance of organismal and cellular homeostasis are considered relevant therapeutic targets for tumor treatment. 1 9 Although apoptosis and autophagy play an important role in the modulation of tumor therapeutics, their molecular action can be counteracted by cancer protective mechanisms. 10 12
Apoptosis is a tightly regulated set of cellular events which are associated with biochemical and morphological changes. However, these characteristic changes of apoptosis present a set of potential targets for cell death and thereby are considered a prognostic marker for early assessment of the efficiency of anticancer agents. Thus, in vivo, apoptosis has been reported to be a relevant detector for tumor resistance and response during the course of the treatment with chemotherapeutics. 13 Autophagy, which is known to be a catabolic pathway, can be activated by cellular stress in response to environmental changes, chemical agents, starvation, or infection. 14 However, the promotion of autophagic process, in response to extra- and intracellular stress, leads to the regulation of different mechanisms that in turn mediate cellular events such as cellular adaptation, cell survival, and cell death.15,16 Therefore, understanding the molecular mechanisms implicated in the regulation of either cell survival or cell death will help to identify a relevant target for tumor prevention and treatment. As a tumor suppressive mechanism, autophagy has been discussed in several studies, in which the distribution of autophagic machinery was found to trigger both cellular transformation and tumor progression.17,18 Additionally, pro- and anti-tumor functions of autophagy have been reported. In addition to its tumor suppressive actions, autophagy can enhance tumor progression of tumor cells exhibiting radio- and chemotherapeutic resistance. 19 The resistance of cancer cells to available therapeutics is thought to be mediated mainly via the activation of autophagy-dependent pathways or via the suppression of apoptosis-dependent pathways. 2 Thus, functional analysis of the mechanisms of tumor resistance to apoptosis in the context of developing novel cancer therapeutics is thought to be an enormous challenge for researchers and clinicians. Additionally, understanding the molecular mechanisms of the crosstalk between apoptosis and autophagy will help to develop new drugs based on their ability to function as BH3 mimetics, a strategy to trigger autophagy-associated cell death in cells conferring resistance to apoptosis. 20 23 While the suppression of apoptosis is linked to the induction of autophagy,2,24 the inhibition of autophagy can cause apoptosis.2,25,26 Thus, identifying the mechanisms thought to be involved in the regulation of the crosstalk between apoptosis- and autophagy-associated cell death is considered an important step for the development of optimal chemotherapeutic approaches for tumor treatment.
Apoptosis
Apoptosis is a form of cell death, also referred to as programmed cell death, in which a ‘suicide’ machinery is activated within the cell, leading to the fragmentation of DNA, shrinkage of the cytoplasm, membrane changes, and finally to cell death without any lysis or damage to neighboring cells. 27 Thus, apoptosis is a normal phenomenon occurring frequently in a multicellular organism, thereby playing an essential role in organism survival. Apoptosis is considered a compelling aspect of various cellular processes. The regulation of apoptosis is mediated by an intracellular proteolytic cascade.1,2,28,29 The mechanisms of this cascade are similar in all animal cells30,31 and depend on a family of proteases that possess a cysteine at their active site with the ability to cleave their target proteins at specific aspartic acids. 32 36 Accordingly, their target proteins are referred as caspases, which are synthesized in the cell in an inactive form (pro-caspases); their activation is mediated by the cleavage of aspartic acids by other caspases. This downstream activation pathway amplifies the intracellular proteolytic cascade. Some of these activated caspases can cleave other key proteins in the cell, such as the nuclear lamins that mediate irreversible breakdown of the nuclear lamina.28,29 Other caspases have been shown to cleave proteins that functionally govern the DNA-degrading enzyme, DNAase, in an inactive form, a cytoprotective mechanism to avoid DNA damage from freeing DNAase and subsequently prevent the initiation of apoptosis. 37 39
Mechanisms of Apoptosis
The pathways, which are involved in the regulation of apoptosis, are extremely complicated and appear to be both agent-dependent and tissues-specific. 40 42 Thus, based on the inducer and the effector, the apoptotic pathways are either extrinsic or intrinsic pathways. 43 45 Besides the major apoptotic pathways, namely, a third apoptotic pathway has been reported. This pathway is specific to cytotoxic T lymphocytes and natural killer (NK) cells and is referred as the perforin/granzyme-A or B pathway.46,47 This perforin pathway is caspase-independent and is mediated through single-stranded DNA damage. 48 Although the variation of their induction manner is great, both extrinsic and intrinsic pathways converge by apoptosis through the activation of caspase-3/7.49,50 An outline demonstrates the molecular mechanisms, which are responsible for the regulation of both extrinsic and intrinsic pathways of apoptosis, are shown (Fig. 1).
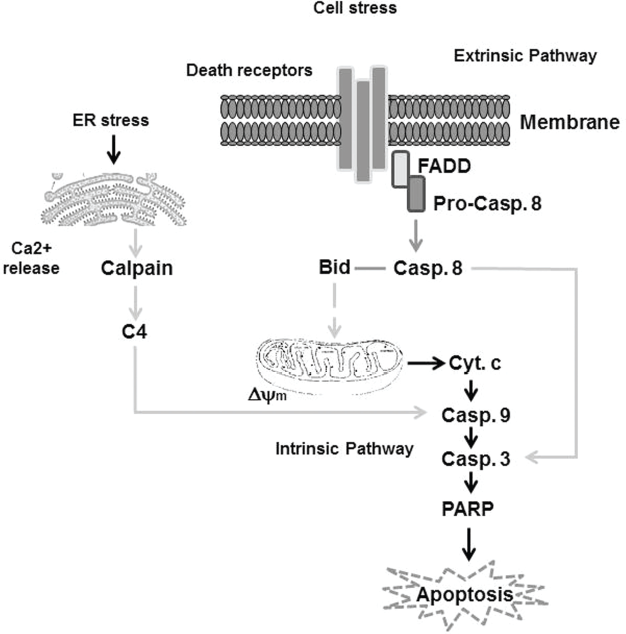
Simplified outline showing key proteins within the extrinsic and intrinsic apoptotic pathways in response to extra- and intracellular stress. Generally, death domain-containing receptors including CD95 (APO-1/Fas) can be activated in response to external signals (such as Fas ligand), which triggers the activity of the extrinsic apoptosis pathway through the Fas-associated death domain (FADD). This pathway is mediated by recruitment and activation of caspase-8 (Pro-casp 8), an initiator caspase, in the death-inducing signaling complex followed by direct cleavage of downstream effector caspases such as caspase-3 (casp 3). Thus, casp 8 is an important pro-apoptotic protein for the extrinsic apoptotic pathway. Initiation of the intrinsic apoptosis pathway results from intracellular stress, leading to mitochondrial damage that is characterized by the loss of mitochondrial membrane potential (Δψm) and cytochrome c (cyt c) release. This triggers activation of initiator caspase-9 (casp 9) and ultimately results in activation of effector caspases, such as caspase 3 (casp 3), that mediates the cleavage of poly(ADP-ribose) polymerase (PARP) and finally leads to apoptosis. Additionally, endoplasmic reticulum stress that can be mediated by oxidative stress elevates intracellular calcium (Ca2+) release that subsequently mediates calpain cleavage, leading to the activation of caspase-4 (casp 4), which mediates the cleavage of casp 9, casp 3, and PARP, and finally leads to apoptosis.
Intrinsic Pathway
The intrinsic pathway is a non-receptor mediated pathway and functions only via mitochondria associated mechanisms. 51 This pathway mediates the intra-cellular signals that act directly on targets, within the cell, or on those associated with the mitochondrial dysregulation. 52 The activation of the intrinsic pathway can act in two opposing patterns.53,54 One of these patterns is mediated by the suppression of the inhibitors of death programs such as growth factors, hormones, and cytokines. However, the other pattern is mediated by direct exposure to several environmental changes such as irradiation, toxins, hypoxia, and viral infections.54,55 In contrast, apoptosis mediated by the intrinsic pathway is characterized by alterations of the inner mitochondrial membrane, that in turn leads to the loss of mitochondrial membrane potential (Δψm).1–4,54 The loss of Δψm leads to the release of the pro-apoptotic proteins, which are characteristic of mitochondrial damage; these molecules include cytochrome c (cyt c), Smac/DIABLO, and the serine protease HtrA2/Omi56,57 apoptosis inducing factors (AIF) endonuclease G and caspase-activated deoxy-ribonuclease (CAD). 54 However, the release of these proteins leads to the activation of caspases such as caspase-9 and caspase-3, whose activation is associated with the loss of Δψm;1,2 the mechanism by which the released cyt c mediates apoptosis is regulated by its binding to both Apaf-1 and pro-caspase-9 to form a protein complex known as the apoptosome.58,59 Through apoptosome formation, caspase-9 becomes active to subsequently mediate the activation of caspase-3, leading to PARP cleavage and finally to apoptosis. The mechanism by which Smac/DIABLO and HtrA2/Omi triggers apoptosis is mediated by the inhibition of inhibitor of apoptosis (IAP) activity. 60 62
However, the appearance of AIF, endonuclease G, and CAD proteins is associated with late apoptosis after the cell has decided to die. 54 In this case, trans-location of AIF to the nucleus leads to DNA fragmentation and subsequently to peripheral chromatin condensation,54,63 a process referred to as “stage I” condensation. 64 Additionally, translocation of endo-nuclease G to the nucleus results in nuclear chromatin cleavage to produce oligonucleosomal DNA fragments. 65 In addition, after release from the mitochondria and subsequent cleavage by caspase-3, CAD can translocate into the nucleus where it mediates the fragmentation of the oligonucleosomal DNA, 66 a process known as “stage II” condensation.54,66 The control and regulation of mitochondrial dysregulation occur through Bcl-2 family of proteins, which are known to be responsible for mitochondrial integrity and subsequent caspase activation, which in turn leads to apop-totic cell death. 67 In this context, the tumor suppressor protein p53 has been widely reported to play a critical role in the regulation of Bcl-2 proteins. 68 Bcl-2 proteins include both pro-apoptotic and anti-apoptotic mediators. 69 These anti-apoptotic mediators include Bcl-2, Bcl-x, Bcl-XL, Bcl-XS, Bcl-w, BAG, Mcl-1, whereas the pro-apoptotic mediators include Bcl-10, Bax, Bak, Bid, Bad, Bim, Bik, and Blk. Thus, the cellular decision to live or to die is controlled by the balance between both pro- and anti-apoptotic mediators, which can regulate the mitochondrial outer membrane permeabilization (MOMP) and subsequently the regulation of cyt c release from the mitochondria into the cytoplasm. 49
The role of Bax and Bak in the regulation of mitochondrial damage has been documented in several studies.1,3 Although the molecular mechanism of both BAK and BAX activation is not well-characterized, the role of Bax has been reported. 69 73 However, phosphorylation of both BAK and BAX serves to facilitate their homo-oligomerization and subsequently their localization into mitochondria, leading cyt c release.74,75 However, the involvement of caspase-8 in the activation of Bid-induced activation of Bax as observed in Fas-induced apoptosis76,77 provides an evidence for the “crosstalk” between the death-receptor (extrinsic) pathway and the mitochondrial (intrinsic) pathway.4,50,76 Additionally, the role of Bad in the promotion of mitochondrial dysregulation has been reported. Thus, phosphorylation of Bad on the serine residue enhances the ability of the 14-3-3 protein, a member of a family of multifunctional phosphoserine binding molecules, to capture Bad in the cytosol where it is sequestered. 78 However, dephosphorylation of Bad or even its native form can translocate into the mitochondria to cause cyt c release. 78 Heterodimerization of Bad with Bcl-Xl or Bcl-2 can lead to enhanced apoptosis by a mechanism that includes neutralizing of the protective effect of both Bcl-Xl or Bcl-2. 79 The main function of both Bcl-2 and Bcl-Xl is to inhibit the release of cyt c from the mitochondria and protect the mitochondria from the localization of pro-apoptotic mediators is not well-characterized. However, the mechanism by which both Bcl-2 and Bcl-XL mediate the inhibition of apoptosis is regulated by the activation of caspase proteases.80,81
Both Puma and Noxa are two members of the Bcl-2 family, which are also involved in the promotion of apoptosis in different cell types including melanoma cells.2,82 Puma plays an important role in p53-mediated apoptosis, and its overexpression, in vitro, is correlated with increased expression, conformational change, and translocation of Bax to the mitochondria, that in turn, lead to the loss of Δψm and subsequently the release of cyt c.2,4,82,83 Noxa has also been reported to be a mediator of p53-induced apoptosis and has been shown to localize to both mitochondria and the endoplasmic reticulum (ER); 82 its interaction with anti-apoptotic Bcl-2 family members mediates caspase-9 activation. 84 Additionally, the regulation of both Puma and Noxa expression by p53 suggests a potential role for both proteins in modulating apoptosis associated with DNA damage. 49 Moreover, the potential role of the proto-oncoprotein Myc in modulating apoptosis via p53-dependent and independent mechanisms has been reported. 85 Therefore, further elucidation of these pathways should provide new insight into the course of tumorigenesis and tumor therapy.
Extrinsic Pathway
The extrinsic pathway is one of the two major apop-totic pathways whose activation is initiated by trans-membrane receptor(s) through the ligation to the corresponding ligand(s) or agonist(s) of interest. These receptors include FasL/FasR, TNF-α/TNFR1, Apo3L/DR3, Apo2L/DR4, and Apo2L/DR5. 86 91 However, one of the most well-characterized receptors includes the member of the tumor necrosis factor (TNF) receptor gene superfamily. 87 These family members share similar cysteine-rich extracellular domains in addition to a cytoplasmic death domain. 87 The main function of the death domain is to transmit the external death signal from the cell's surface to the intracellular signaling pathways. Following binding of the ligand to the corresponding receptor, cytoplasmic adapter proteins can exhibit the corresponding death domains. Ligation of Fas ligand to the Fas receptor mediates its binding to FAS-associated death domain (FADD), whereas ligation of the TNF ligand to the corresponding TNF receptor mediates its binding to TNF-receptor associated death domain (TRADD), which in turn leads to recruitment of FADD and receptor-interacting protein (RIP).92,93 The interaction of FADD with pro-caspase-8 through its death effector domain leads to formation of a death-inducing signaling complex (DISC) that mediates the activation of caspase-8. 94 Following cas-pase-8 activation, death receptor-mediated apoptosis can be initiated.
Execution Phase
Both extrinsic and intrinsic pathways lead to the final pathway of apoptosis. 49 The execution phase includes activation of execution caspases, which are characteristic of the final phase of apoptosis that is associated with the activation of cytoplasmic endonucleases and proteases. This leads to the degradation of nuclear materials and cytoskeletal proteins.
Both morphological and biochemical changes characteristic of apoptotic cells are mediated by effector “executioner” caspases such as caspase-3, caspase-6, and caspase-7. Activation of these caspases results in cleavage of various substrates such as cytokeratins, PARP, plasma membrane cytoskeletal protein alpha fodrin, and nuclear mitotic apparatus protein (NuMA). 95
Caspase-3 is known to be the most important cas-pase among the executioner caspases and its activation is mediated by initiator caspases such as caspase-8, caspase- 9, or caspase-10. 77 In apoptotic cells, activation of caspase-3 mediates activation of various substrates such as endonuclease caspase-activated DNase (CAD), whose activation promotes protein degradation that subsequently causes chromatin condensation in apoptotic cells. 96 Additionally, activation of keratin 18 by caspase-3 mediates formation of apoptotic bodies during the course of apoptosis. 97 These reports, however, describe the mechanisms responsible for caspase-3-mediated disruption of the cytoskeleton during apoptosis. Moreover, caspase-cleaved gelsolin functions as actin filaments in vitro in a Ca2+-independent manner. 98 Expression of the gelsolin cleavage product in multiple cell types triggers the cells to become round, detach from the plate, and undergo nuclear fragmentation, 99 suggesting an important role for activated caspase-3-induced cleavage of gelsolin in apoptosis regulation.
After the execution phase is complete, unwanted cellular components should be removed. Therefore, the last step of apoptotic process involves phagocytic uptake of apoptotic cells. 49 Phospholipid asymmetry and externalization of phosphatidylserine on the surface of apoptotic cells and membrane fragmentation are characteristic of the execution phase. 49 Although the mechanisms responsible for regulating phosphatidylserine translocation to the outer leaflet of the cell has not been described in detail, translocation of phosphatidylserine to the outer side is thought to be mediated by the loss of aminophospholipid translocase activity and nonspecific flip-flop of phospholipids of various classes. 100 Accordingly, the roles of Fas, caspase-8, and caspase-3 in regulating phosphatidylserine externalization has been demonstrated.101,102 Although caspase-independent phosphatidylserine exposure was reported to occur during apoptosis of primary T-lymphocytes, 101 phosphatidylserine externalization in response to oxidative stress has been reported in erythrocytes. 102 Moreover, externalization of phosphatidylserine in the execution phase appears to be an essential cellular mechanism for removing apoptotic cells since the appearance of phosphotidylserine on the outer leaflet of apoptotic cells may facilitate non-inflammatory phagocytic recognition, thereby allowing their early uptake and finally their removal. 103
Autophagy
Autophagy is a highly conserved degradation pathway that discards damaged cellular components and is morphologically characterized by the formation of double membrane autophagosomes.2,51 Sequestration of impaired organelles or unwanted cellular components by autophagy facilitates their delivery to lysosomes for degradation and recycling. 104 106 In addition to eliminating damaged cellular components, autophagy has been implicated in several physiological and pathological processes, including cell survival and cell death. 107 Additionally, the involvement of autophagy in cellular homeostasis and cell and tissue renovation has been reported. 108 111 More importantly, the participation of autophagy in the etiopathogenesis of many important human diseases, including metabolic disorders and cancer, has been reported. 112 Autophagy has been demonstrated to function as an important cell survival mechanism, particularly when cells are under stress conditions such as oxidative stress and starvation. 113 115 Additionally, the involvement of autophagy in regulating cell death processes, in apoptosis,2,116,117 necrotic cell death,118,119 or even autophagic cell death, has been reported. 120 122 Thus, the function of autophagy as cell death machinery in different tissue types appears to be established. However, the factors responsible for regulating this mechanism are not well-understood. Although the natural and cellular stress-induced autophagy is crucial processes, the factors responsible in determining whether autophagy mediates cell survival or cell death remain unknown.
Mechanisms of Autophagy
Despite its role in maintaining intracellular integrity, 123 autophagy is a tightly regulated process that is regulated by distinct cellular mechanisms. One of these mechanisms is the target of rapamycin (TOR) kinase that serves as a control point downstream of signaling responsible for different cellular events such cell growth, metabolism such as growth factor receptor signaling, and insulin signaling.124,125 It was previously shown that activation of TOR kinase is mediated by Akt kinase, PI3-kinase, and growth factor receptor signaling in response to the availability of nutrients to promote growth via induction of ribosomal protein at both transcriptional and translational levels. 126 Thus, under cell growth conditions, the TOR pathway can mediate the inhibition of autophagy by suppressing Atg1 kinase activity. 127 Additionally, under low levels of adenosine-5'-monophosphate (AMP) or under hypoxia conditions, TOR kinase can be repressed, which blocks the activity of the tumor suppressor proteinsTsc1/Tsc2 as mediated via Rheb, a small GTase required for mTOR activity.126,128 Reduction of Akt activity through inhibition of growth factor receptor or by rapamycin are thought to be possible mechanisms, which are responsible for the repression of TOR kinase. 129 Therefore, inhibiting the TOR kinase that is mainly involved in regulating autophagy in response to altered physiological conditions is mediated by the inhibition of growth factor receptors or by inhibitors, leading to increased catabolism, which is considered to be a therapeutic strategy for tumor treatment.
Accordingly, rapamycin and its derivatives, which are known to inhibit mTOR function through a kinase-independent mechanism, have been tested in clinical trials in several tumor types.130,133 However, the action of this inhibitor is mediated by a kinase-independent mechanism that inhibits tumor growth via suppression of protein translation machinery and induction of autophagy. 130 In addition to its role in regulating tumor progression and autophagy, TOR has been reported to function as the catalytic component of two distinct complexes, including TORC1 and TORC2. 131 Hypoxia can also activate autophagy by hypoxia inducing factor (HIF)-dependent 132 or independent mechanisms. However, these mechanisms are regulated via the inhibition of TOR mediated by adenosine monophosphate kinase (AMPK), REDD1, and Tsc1/Tsc2.129,133 Moreover, the most specific targets of autophagy include BNIP3 and BNIP3L; the BNIP3 subfamily of BH3-only proteins can form stable homodimerization complexes that localize to the outer membrane of the mitochondria after cellular stress.134,135 Although these proteins are linked to cell death, their main function is implicated in the regulation of autophagy.134,135 Thus, the main role of BNIP3L/NIX in autophagy is to mediate mitochondrial clearance during the maturation of reticulocytes. 136 139 Although the functional role of BNIP3 and BNIP3L in regulating both autophagy and cell death is not well-characterized in detail, various models have been proposed to explain the functional role of BNIP3/BNIP3L in autophagy. 140 One of these models describes the role of BNIP3 in deregulation of Beclin-1 by disrupting its interaction with Bcl-2, 141 whereas other models of the interaction between BNIP3 and Rheb have been discussed in the context of hypoxia-induced autophagy. 142
Besides its role in death and cell survival, autophagy is also involved in regulating cell cycle arrest. However, the mechanism responsible for modulating autophagy-induced cell cycle arrest is thought to be regulated by inhibition of the TOR pathway in response to nutrient deprivation by inhibiting the translation machinery of key cell cycle genes, such as cyclin D1. 143 The possible mechanisms, which are responsible for the regulation of autophagy in mammalian cells, are outlined in Figure 2.
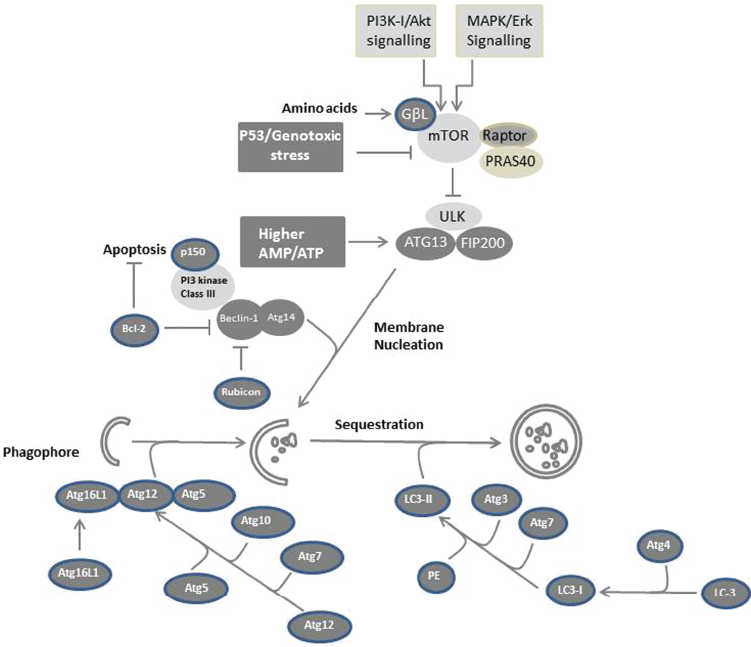
Possible mechanisms of autophagy in mammalian cells. The formation of autophagosomes is mediated by a conserved pathway in all mammalian cells in response to variable cellular stresses. Cellular stress that results from the availability of nutrients (starvation) or from the exposure to growth factors such as endothelial growth factor (EGF) lead to activation of PI3K-I/Akt and MAPK/ERK signaling, respectively. ULK-Atg13-FIP200 complexes mediate mTOR signaling downstream of the autophagy machinery. Starvation or growth factor treatment suppresses mTOR-mediated phosphorylation of ULK and Atg13 resulting in ULK-mediated phosphorylations of Atg13, FIP200, and ULK itself, and subsequently to membrane nucleation. Downstream of the TOR-suppressive signaling pathway, Atg proteins function to form the autophagosome. The PI3 kinase complex (class III) including Beclin-1/Atg6 is required to initiate autophagosome formation. Elongation of the isolation membrane requires Atg12 and Atg8 (LC3) ubiquitin-like modification systems. A ubiquitin-like protein, Atg12, is conjugated with Atg5 and forms a large complex with Atg16L1. LC3, a ubiquitin-like protein, associates with both the isolation membrane and the completed autophagosome as a conjugate with phosphatidylethanolamine.
Crosstalk between Apoptosis and Autophagy
Based on their central role in cell survival and cell death, mitochondria have been shown to play an essential role in the regulation of apoptosis in response to extensive cellular stress leading to the loss of Δψm, and to the subsequent release of pro-apoptotic molecules such as cyt c, 2 4 and AIF. 1 The accumulation of both cyt c and AIF in the cytoplasm results in the activation of caspases and, finally, apoptotic cell death.1,2 However, if the loss of Δψm is limited to only a subset of mitochondria, selective autophagosomal machinery will eliminate the limited subset of depolarized mitochondria, a cellular function that serves as cytoprotective mechanism to prevent mitochondrial damage. 144
Apart from their molecular authenticity and induction manner, the involvement of several proteins that regulate both apoptosis and autophagy has been reported. 145 Accordingly, the anti-apoptotic protein Bcl-2 has been shown to play an essential role in regulating both autophagy and apoptosis as mediated by the binding of the pro-autophagic protein Beclin1 with pro-apoptotic proteins such as Bax. Thus, extensive cellular stress can lead to the loss of Δψm that mainly results in the release of mitochondrial proteins such as cyt c and AIF and subsequently initiates apoptosis.1,146,147 Additionally, the depletion of nutrients leads first to the release of Beclinl, that in turn induces activation of PI3K and subsequently the induction of autophagy.148,149 The extension of nutrient deprivation can lead to release of the pro-apoptotic protein Bax from Bcl-2, which mediates the loss of Δψm and subsequently initiates the apoptotic machinary.116,150 Thus, the molecular coupling of both autophagy and apoptosis is determined by the type and longevity of cellular stress.151,152 Additionally, the pattern and fashion of the subcellular localizations of Bcl-2 at the ER and/or to mitochondria is thought to be the main factor that determines the switch between autophagy and apoptosis.153,154 However, the role of Beclin-1 in regulating autophagy is mediated by the by localization of Bcl-2 at the ER.155,156 The regulation of apoptosis can be mediated through the localization of Bcl-2 to mitochondria. 157 Accordingly, the cellular outcome between autophagy or apoptosis appears to depend on the entity of the mitochondria rather than the molecular mechanism induced.1,2 Therefore, mito-chondrial damage can overpower the pro-survival autophagic pathway, leading to apoptosis via a mechanism mediated by the release of pro-apoptotic proteins such as Bax from anti-apoptotic proteins such as Bcl-2. Moreover, the involvement of a number of proteins in regulating the crosstalk between autophagy and apoptosis has been reported. These include the autophagy-regulating proteins Beclin1, the class III PI3K, and ATG4D. 158 160 Thus, the cleavage of these proteins by caspases facilitates their localization to mitochondria where they serve new functions such as promoting mitochondria-mediated apoptosis. Therefore, the destruction of autophagic function of Beclin-1 and PI3K through caspase-dependent cleavage is considered to be an alternative strategy for tumor treatment. The autophagy-regulating protein 5 (ATG5) is a member of autophagy-regulating protein family. This protein is characterized by its ability to link apoptosis and autophagy. 161 Upon autophagy induction, ATG5 becomes active and subsequently initiates autophagosomic formation. 161 Additionally, cleavage of ATG5 by calpain in response to cellular stress promotes localization of cleaved ATG5 products to the mitochondria, where they bind to Bcl-XL, leading to the loss of Λψm, and finally to apoptosis. 162 164 Thus, unlike cleaved Beclin1, class III PI3K, or ATG4D, cleaved ATG5 appears to functionally trigger apoptosis without any additional apoptotic mediators.
Although the functional role of Beclin-1 as Bcl-2-interacting protein, in addition to be an initiator factor for the development of autophagic formation, 165 its precise role in regulating the crosstalk between autophagy and apoptosis has not completely characterized. The anti-apoptotic protein Bcl-2 is the most thoroughly described prototypic member of the Bcl-2 family of apoptosis-regulating proteins, 165 in addition to being characteristic for their Bcl-2 homology (BH) domains. 165 Thus, based on the number of functions of these BH domains, three groups are recognized. One of these groups includes the multi-domain anti-apoptotic Bcl-2 family proteins such as Bcl-2, Bcl-xL, Bcl-w, A1, and Mcl-1, the second one includes the multi-domain pro-apoptotic Bcl-2 family proteins such as Bax, Bak, and Bok, and the third group includes pro-apoptotic Bcl-2 family members that possess only the BH3 domain and are therefore referred to as BH3-only proteins such as Bim, Bid, Bad, Bmf, Puma, Noxa, Bik, Hrk, and Mule. The main function of these multidomain pro-apoptotic Bcl-2 family members (Bax and Bak) is to induce the loss of Δψm that subsequently leads to the release of cyt c and, in turn, the activation of downstream caspases such as caspase-3 and 9.1–3,76,82 Anti-apoptotic Bcl-2 family members serve mainly to inhibit pro-apoptotic proteins, such as Bax and Bak. 166 Despite the similarity of their pattern and their function, the manner and function of the BH3-only proteins in modulating their anti-apoptotic function is different. Some BH3-only proteins exert their function by their binding to the anti-apoptotic Bcl-2 family members, and thereby block their function as repressor for the multi-domain of the pro-apoptotic Bcl-2 proteins.167,168 Otherwise, these BH3-only proteins exert their function by the direct interaction with the multi-domain of pro-apoptotic proteins and thereby block their pro-apoptotic function.169,170
In addition its function as interacting partner of Bcl-2, Beclin-1 has been reported to play an important role in antiviral host defense in some studies, 165 whereas in another study it was shown to function as a tumor suppressor protein. 171 In other studies, Beclin-1 has been reported to function as an ortholog of the yeast autophagy protein Atg6 and thereby is implicated in the regulation of autophagic formation in mammalian cells. 172 Accordingly, the expression of Beclin-1 was found to be essential for promoting autophagic formation in Atg6-deficient yeast. 172 Additionally, reduction of the tumorigenicity of Beclin-1- expressing cells confirmed further the antitumor function of Beclin-1. 172 Functional analysis of Beclin-1 suggested a molecular mechanism whereby Beclin-1 initiates autophagy.173,174 These mechanisms appear to be mediated through the interaction of Atg6/Beclin-1 with the phosphoinositide 3-kinase (PI3K).173,174
However, the identification of Beclin-1 as a Bcl-2 binding partner revealed that the interaction between Bcl-2 and Beclin-1 is involved in the formation of a complex that is thought to play a central role in modulating the crosstalk between signaling pathways of apoptosis and autophagy.175,176 As with other BH3-only proteins, Beclin-1 was shown to interact with Bcl-2, Bcl-xL, and Mcl-1 through the BH3 domain; evidence for this is the inhibition of the observed interaction in response to mutations in BH3 domain or even in the receptor domain in anti-apoptotic Bcl-2 family members. 175 176
More importantly, the interaction between Beclin-1 and the BH3 domain of anti-apoptotic Bcl-2 family members is thought to be the primary mechanism responsible for the inhibition of Beclin-1- mediated induction of autophagy under nutrient-adequate conditions.177,178 Although the Bcl-2, Bcl-xL, and Mcl-1 proteins are mainly localized to the mitochondria as a protective mechanism for mitochondria, the localization of Bcl-2 family members to the ER has been shown to inhibit starvation-induced autophagy.177,178 Although extensive studies on the role of Bcl-2 family members have examined inhibition of the autophagic function of Beclin-1, the precise role of Beclin-1 in the processes of autophagic formation is not fully understood. However, the identification of the novel Bcl-2 interacting partner, nutrient-deprivation autophagy factor-1 (NAF-1), 179 provided insight into the mechanism of the interaction of Bcl-2 with Beclin-1. This provided insight into the role of Beclin-1 in the process of autophagic formation. Accordingly, the function of NAF-1 appears to be implicated in the stabilization of the Bcl-2-Beclin-1 complex at the ER; 179 therefore, the loss of NAF-1 is responsible for disrupting the Bcl-2-Beclin-1 complex and subsequently inducing autophagy, which is evidence for the inhibitory role of Bcl-2 family members in Beclin-1-mediated autophagy. 179
The mechanism by which Beclin-1 mediates the induction of autophagy under starvation or stress conditions is thought to be regulated by the release of Bcl-2 and Bcl-XL from the formed complex with Beclin-1.178,180 Therefore, the dissociation of Bcl-2/Bcl-XL-Beclin-1 has been reported to be regulated by c-Jun-N-terminal kinase-mediated phosphorylation of Bcl-2 181 184 death-associated protein kinase (DAPK)-mediated phosphorylation of Beclin-1, 188 190 translocation of the nuclear protein high-mobility group box 1 (HMGB1) to the cytosol,188,189 or competition with other BH3-only proteins for Bcl-2 binding.180,185,186,188–190
Induction of mitogen activated protein kinase (MAPK), c-Jun-N-terminal kinase (JNK), in response to cellular stress results in the phosphorylation of the three residues in the regulatory loop of Bcl-2, leading to disruption of its binding to Beclin-1, 180 an essential step in the induction of autophagy in nutrient-shortage conditions. Thus, cells with either mutations in the phosphorylation sites of the regulatory loop of Bcl-2 or even deficiency in JNK activation appear to be unable to undergo starvation-induced autophagy. 180 Therefore, the constitutive activation of JNK can induce autophagy under nutrient-sufficient conditions in Bcl-2-expressing cells. 180 The involvement of JNK in modulation of autophagy in response to ER stress, oxidative stress, cancer drugs, and stimulation also occurs through the death receptor Fas. 181 184
In addition, the disruption of Bcl-2-Beclin-1 complex is thought to be a target for pro-apoptotic BH3-only proteins such as Bad and Bax, which can mediate the inhibition of Beclin-1-mediated induction of autophagy.187,190
Although the essential role of Beclin-1 in the link between autophagy and apoptosis has been reported in several studies, complex formation of Beclin-1/Vps34 is not necessary for the modulation of autophagic formation in all cell types. 191 194
Besides the ability of Bcl-2 family proteins to regulate autophagy through the interaction with Beclin-1, caspases have been shown to inhibit autophagy via a mechanism mediated by the cleavage of autophagy-related proteins.2,195 Accordingly, the role of apoptosis-associated proteases in the regulation of the balance between apoptosis and autophagy has been reported. 164 Additionally, the detection of N-terminal cleavage products of Atg5 in multiple cell types and its translocation to the mitochondria leads to mito-chondrial dysregulation and subsequently induction of cyt c release 164 suggests an essential role for caspases in the inhibition of autophagy. More importantly, the promotion of apoptosis by overexpression of N-terminal Atg5 cleavage products is considered evidence for the ability of the cleaved Atg5 to trigger apoptosis. 164 Although N-terminal cleavage products of Atg5 are unable to promote autophagy, 164 the pro-apoptotic function of Atg5 seems to be limited and can perhaps sensitize only tumor cells to anti-cancer agents-induced apoptosis. 164 Moreover, overexpression of LC3 did not appear to exert any effect on apoptosis since the enhanced sensitivity to apoptosis observed in cells expressing cleaved Atg5 is linked to the pro-apoptotic function of the N-terminal Atg5 fragment 164 rather than to enhanced autophagy.
In addition to the ability of apoptosis-associated proteases in regulating the balance between apoptosis and autophagy, a link between the upstream signals that induce apoptosis through the extrinsic pathway has been reported. 196 Extrinsic apoptosis is initiated by the ligation of death receptors with their cognate ligands, which results information of a death-inducing signaling complex (DISC) from Fas-associated protein with the death domain (FADD) and the initiator caspase, pro-caspase-8. Formation of this complex results in the cleavage of pro-caspase 8 and subsequently the release of active caspase 8, which cleaves and activates downstream targets, including effector caspases.197,198 Thus, in addition to its roles in regulating apoptosis, a key component of DISC has been also reported in several studies to be involved in modulating autophagy. 199 201 Additionally, the deficiency or even the inhibition of caspase-8 was found to result in excessive autophagic formation in different cell types. 199 Moreover, mutations in FADD were found to prevent its interaction with pro-caspase-8, resulting in the inhibition of apoptosis and a switch to autophagy.200,201
Although Atg5 has been shown to interact with FADD through its death domain (DD), the deficiency of FADD does not appear to influence autophagic formation. 202 In contrast, the loss of FADD was found to recover the increased caspase-dependent cell death induced by overexpression of Atg5, evidence for the important role of the Atg5-FADD interaction in regulating apoptosis rather than autophagy. 202
Interestingly, the induction of autophagy in cells by the inhibition of extrinsic apoptotic pathways is thought to be associated mostly with an increased induction of cell death.202,203,205 Although the role of Atg5 is essential for both autophagosomic formation and cell death by exposure to IFN-y, the ability of N-terminal cleavage product of Atg5 to induce apoptosis indicates that Atg5-mediated cell death is not associated with the loss of the autophagic function of Atg5.164,202 The possible pathways, which are thought to be involved in the modulation of the cross talk between apoptosis and autophagy, are outlined in Figure 3.
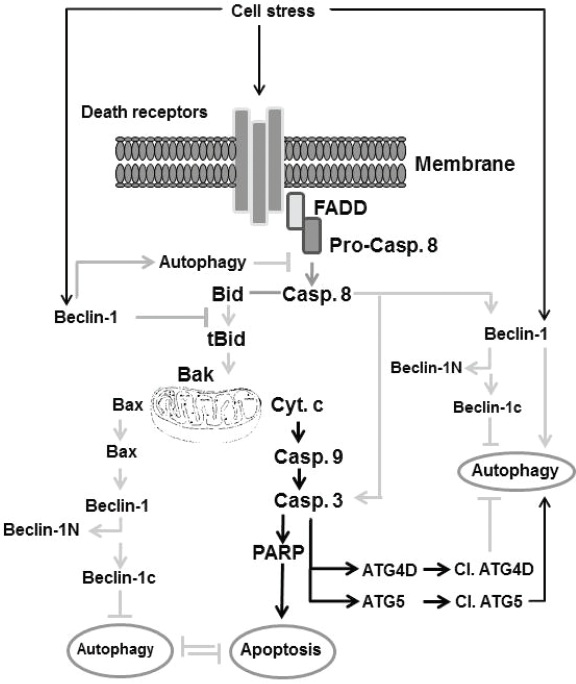
Outline of the molecular mechanisms thought to be involved in the regulation of the cross-talk between apoptosis and autophagy. Some extracellular stress can trigger both apoptosis and autophagy via receptor-dependent and independent mechanisms. One of these mechanism includes the induction of Beclin-1 that triggers autophagy, that in turn inhibits apoptosis by a mechanism mediated by the inhibition of caspase-8 cleavage, and subsequently blocks the cleavage of Bid. Another mechanism includes the activation of death receptor, leading to cleavage of caspase-8 via the Fas-associated death domain. The cleavage of caspase-8 leads to cleavage of Bid, which triggers the loss of mitochondrial membrane potential characterized by cyt c release, caspase-9, caspase-3, and PARP cleavage, the hallmark of apoptosis. Cleavage of caspase-3 triggers cleavage of ATG40, leading to the inhibition of autophagy, whereas the cleavage of ATG5 by caspase-3 results in induction of autophagy. Additionally, accumulation of phosphorylated Bax triggers cleavage of Beclin-1 into Beclin-1N and Beclin-1c. Accumulation of Beclin-1N leads to inhibition of induced autophagy.
Therapeutic Strategies
Based on intensive studies focusing mainly on the modulation of autophagy through apoptotic signaling pathways, it is becoming clear that autophagy is involved in the regulation of apoptosis. Thus, the inhibition of apoptosis by autophagy has been shown to be regulated by the degradation of pro-apoptotic proteins including caspases. 203 More important, autophagy is involved in regulating cancer development and progression in addition to being a key factor for determining tumor cell sensitivity to anticancer therapy.204,205 Accordingly, several conventional and experimental antitumor strategies have been reported as relevant strategies for tumor therapy. These strategies include the combination of conventional therapies with autophagy regulators.204,205 These autophagic regulators include rapamycin,206,207 arsenic trioxide,208,209 temozolomide,210,211 kringle domains of plasminogen, 212 phenethyl isothiocyanate, 213 OSU03012 (derivative of celecoxib), 214 NVPBEZ235,215,216 and cell wall skeleton of Mycobacterium bovis Bacillus Calmette Guerin. 217 Additionally, other autophagic regulators such as phenethyl isothiocyanate, the celecoxib derivative OSU03012 and kringle domains of plasminogen (endostatin kringle 5, K5) have emerged as promising cancer chemopreventive agents based on their ability to induce autophagic cell death. 213 Moreover, several cancer therapies, including DNA-damaging chemo-therapeutic temozolomide 218 and radiation 219 have been shown to induce autophagy both in vitro and in vivo. 220 However, this induced autophagy has been shown to serve as a protective mechanism against the toxicity of the applied anticancer agents. 221 Additionally, radiation therapy has been reported to promote autophagy by a mechanism mediated through the upregulation of autophagy mediators including Beclin-1, ATG3, ATG4, ATG5, and ATG12.219,220 Furthermore, some chemotherapeutic agents such as histone deacetylase inhibitors 221 and cisplatin 222 have been shown to induce autophagy through the accumulation of reactive oxygen species in mitochondria. Preclincal studies have also addressed a powerful therapeutic potential for curcumin in tumor treatment; however, the therapeutic potential of curcumin due the ability of curcumin to trigger both apoptosis 223 and autophagy. 224
The reliability of proteasome inhibitors alone or in combination with other drugs has been reported to induce apoptosis as well as autophagy in different tumor types including melanoma.2,225–227 However, the induction of apoptosis by proteasome inhibitors is linked to the suppression of transcription of anti-apoptotic factors 228 230 and is associated with the upregulation of pro-apoptotic proteins.2,225–227 While induction of autophagy by proteasome inhibitor regulated by the stabilization of anti-apoptotic proteins including the protein Mcl-1, as shown in melanoma cells. 2
Based on previous and current findings, autophagy and apoptosis appear to play opposing roles in cancer cells since the inhibition of autophagy by chloroquine was found to sensitize tumor resistance to anti-cancer agents-induced apoptosis. 231 234 Additionally, the role of autophagy in the resistance of tumor cells to the pro-apoptotic effects of TRAIL has been reported in several studies. 234 236 Unlike most superfamily members, TRAIL as a cancer therapeutic possesses pro-apoptotic effects that is specific to tumor cells.237,238 However, the advantage of TRAIL as a tumor therapeutic agent becomes unattractive based on the development of TRAIL resistance in many tumors. 239 Therefore, intensive studies on the tumor resistance mechanisms of TRAIL revealed that the failure of TRAIL to trigger apoptosis of tumor cells is associated with the cyto-protective effects of autophagy. Correspondingly, c-FLIP-expression is resistant to TRAIL-induced apoptosis and is characterized by the increase of Beclin-1 expression and a marked autophagosome formation in response to the activation of TRIAL or Fas receptors. 240 Accordingly, the inhibition of the autophagic machinery in cells expressing c-FLIP or in Bax-deficient cells enhances their sensitivity to TRAIL-mediated cell death.234,235 Thus, based on the findings of widely reported studies, apoptosis and autophagy appear to work together. While autophagy mediates the degradation of caspases, caspases can also target the cleavage of autophagy-related proteins.
However, the use of autophagy by tumor cells as a cytoprotective mechanism to block chemotherapeutic agents-induced apoptosis seems to be a general phenomenon. Cytoprotective effects associated with autophagy were reported in different tumor types following treatment with anticancer agents including gefitinib, erlotinib, and celecoxib. 240 242 Despite these ongoing clinical efforts, the use of autophagy inhibitors as a therapeutic strategy in cancer, thus far, has not been established. Therefore, further preclinical evaluation to optimize the efficacy of the suggested therapies is urgently needed.
Taken together, in addition to the elucidation of molecular mechanisms, whereby autophagy counteracts or synergizes apoptosis, or vice versa, understanding of the details of these mechanisms provides insight into the potentials and limitations of the combination of cancer therapies targeting both apoptosis and autophagy.
Conclusion
Although the relationship between apoptosis and autophagy has been widely reported, the identification of the molecular mechanisms which govern this relationship have not been described in detail. Thus, understanding the molecular mechanism of the crosstalk between apoptosis and autophagy may help to develop a relevant therapeutic strategy for tumor treatment. In this review, we provided insight into the molecular mechanisms of the crosstalk between apoptosis and autophagy. In addition, molecular targets, which are thought to be essential for the balance between autophagy and apoptosis, have been discussed. Additionally, the relevant pathways of both apoptosis and autophagy, which are thought to be targets for novel therapeutic approaches, were discussed.
Author Contributions
Conceived and designed the experiments: MH, AE. Analyzed the data: MH. Wrote the first draft of the manuscript: MH, AE, YH, and DS. Contributed to the writing of the manuscript: MH, YH. Agree with manuscript results and conclusions: MH. Jointly developed the structure and arguments for the paper: MH, AE. Made critical revisions and approved final version: MH, YH. All authors reviewed and approved of the final manuscript.
Funding
This work was supported by grants from German Research Foundation (HA 5081/3-1), from L'Alsace Contre le Cancer, France, and from German Cancer Research Foundation (10-2202-Ha 1) to M. Hassan.
Competing Interests
Authors disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests. Provenance: the authors were invited to submit this paper.