
Abstract
Mitochondrial dysfunction occurs in neurodegenerative diseases, however molecular mechanisms underlying this process remain elusive. Emerging evidence suggests that nitrosative stress, mediated by reactive nitrogen species (RNS), may play a role in mitochondrial pathology. Here, we review findings that highlight the abnormal mitochondrial morphology observed in many neurodegenerative disorders including Alzheimer's, Parkinson's, and Huntington's diseases. One mechanism whereby RNS can affect mitochondrial function and thus neuronal survival occurs via protein S-nitrosylation, representing chemical reaction of a nitric oxide (NO) group with a critical cysteine thiol. In this review, we focus on the signaling pathway whereby S-nitrosylation of the mitochondrial fission protein Drp1 (dynamin-related protein 1; forming S-nitrosothiol (SNO)-Drp1) precipitates excessive mitochondrial fission or fragmentation and consequent bioenergetic compromise. Subsequently, the formation of SNO-Drp1 leads to synaptic damage and neuronal death. Thus, intervention in the SNO-Drp1 pathway may provide therapeutic benefit in neurodegenerative diseases.
Keywords
Introduction
Mitochondria are dynamic organelles capable of establishing interconnected networks. Dynamic morphological changes are mediated by continual, highly-regulated fusion and fission events, collectively termed mitochondrial dynamics.
1
For example, the evolutionarily conserved GTPase, dynamin related protein1 (Drp1), is known to participate in the process of mitochondrial fission.
1
When mitochondria undergo fission, cytosolic Drp1 translocates to the outer mitochondrial membrane, oligomerizes around mitochondrial tubules, and in conjunction with Fis1 promotes division or fission.
1
The fusion machinery consists mainly of three additional large dynamin-like GTPases: both mitofusin1/2 (Mfn1/2) and optic atrophy protein 1 (Opa1).
2
The strict regulation of the equilibrium between fusion and fission events governs mitochondrial dynamics in a healthy cell and is crucial to maintaining mitochondrial function affecting energy supply, response to apoptotic stimuli, mitochondrial inheritance via admixture of mitochondrial-DNA (mtDNA), and translocation of mitochondria to regions of high-energy demand.
3
Although mitochondrial dynamics are crucial in all cell types, neurons are thought to be especially dependent on a dynamic mitochondrial network.
4
With their often long processes, neurons require proper distribution of mitochondria across long distances to satisfy the energy requirements of electrical excitability and synaptic transmission (Fig. 1). To this end, healthy neurons precisely control mitochondrial dynamics in order to efficiently transport mitochondria to distal locations, including dendrites, axons, and synapses.
3
Thus, mitochondrial function is essential to many neuronal functions, including energy production, Ca2+ buffering, axonal and dendritic transport, and release and re-uptake of neurotransmitter.3,5
Proposed model of excessive mitochondrial fission contributing to neurodegenerative disorders. Balanced mitochondrial dynamics (mitochondrial fission and fusion events) facilitate proper distribution of neuronal mitochondria within neuronal dendrites and into synaptic termini. In addition, the precise control of mitochondrial fission and fusion events in axons is needed to support normal neuronal function (e.g., via production of ATP and buffering of Ca2+). Malfunction of the fission or fusion machinery can result in excessive mitochondrial fragmentation under neurodegenerative conditions, leading to bioenergetic failure and subsequent synaptic damage.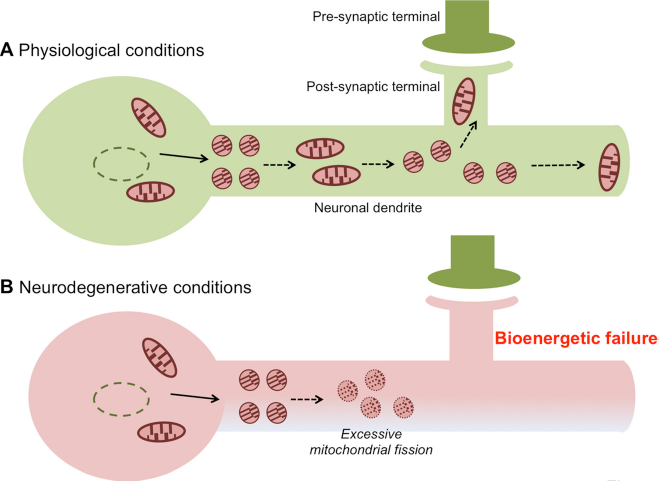
In order to maintain a proper distribution of functional mitochondria, both fusion and fission must occur in homeostatic balance. Basal activity of the fission machinery is required to divide the mitochondrial network into transportable units which can then be efficiently distributed to synapses. 3 Fusion is necessary to maintain bioenergetic integrity of mitochondria. 5 If the balance of fission and fusion becomes biased (e.g., with increased fission or impaired fusion) excessive mitochondrial fragmentation is observed, resulting in impaired mitochondrial translocation and energy production at synaptic sites (Fig. 1).1,3 The synaptic dysfunction thus induced is typically followed by dendritic and axonal degeneration, leading to neurodegeneration. Indeed, excessive mitochondrial fragmentation with damaged cristae and resulting impaired bioenergetics are often associated with many neurological conditions, including stroke, Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD), as well as several metabolic diseases. 5 8 Pathological disturbance of mitochondrial dynamics can result from either: 1) rare genetic mutations, such as those described for Mfn1/2, Opa1 and Drp1, which underlie Charcot-Marie-Tooth neuropathy type 2A (CMT2A), 9 autosomal dominant optic atrophy (ADOA),6,10 and a rare disorder of brain development, 11 respectively; or 2) altered posttranslational modifications (PTMs) of the proteins encoded by these genes that are responsible for the fission/fusion machinery.5,12
In this review, we discuss recent findings concerning aberrant PTMs which regulate the mitochondrial fusion and fission machinery and thus contribute to the pathophysiology of neurodegenerative diseases. In particular, we focus on involvement of nitrosative stress-induced Drp1 activation, which results in excessive mitochondrial fragmentation, impaired bioenergetics, and consequent synaptic damage in neurodegenerative disorders.7,12,13
Mitochondrial Dynamics in Neurodegeneration
As mentioned above, tight regulation of the mitochondrial fission/fusion machinery is essential for maintaining healthy neuronal physiology. However, dysfunctional mitochondrial dynamics caused by impaired mitochondrial fission or fusion is a hallmark of many neurodegenerative diseases and contributes to their pathophysiology.
Impaired Mitochondrial Fission and Fusion in Neurodegenerative Diseases
Mitochondrial fusion is the process by which the inner and outer mitochondrial membranes are separately fused by Opa1 and Mfn1/2, respectively.3,6 Mutations in these genes result in rare neurological conditions. For example, loss of function mutations in the Mfn2 gene disrupts mitochondrial fusion, resulting in distal muscle weakness and sensory loss in CMT2A. 14 To date, more than 40 mutations have been identified, most of which are located in the conserved GTPase domain of Mfn2.9,15 In mouse models, deletion in the mouse Mfn2 gene causes severe mtDNA loss and the accumulation of mtDNA mutations. 16 In addition, Mfn2 deficiency results in decreased axonal transport of mitochondria, causing a lack of functional mitochondria at pre- and postsynaptic sites. 17 Collectively, these findings suggest that Mfn2 mutations in CMT2A patients may trigger defects both in mtDNA integrity and mitochondrial transport, contributing to disease pathogenesis.
Another neurologic disorder caused by disruption in the fusion machinery is ADOA, 6 representing an hereditary optic nerve atrophy characterized by degeneration of retinal ganglion cells and progressive loss of vision. Mutations in the Opa1 gene, and to a lesser extent in Opa3, underlie ADOA. 10 Similar to Mfn2 mutations, over 100 reported mutations in Opa1 target predominantly the GTPase domain. 18 Opa1 is located at the inner mitochondrial membrane, regulating fusion and modeling of the cristae structures. Interestingly, most Opa1 mutations identified in the GTPase domain cause ADOA via a dominant-negative effect. Furthermore, heterozygous Opa1 knockout mice manifest severe degeneration of the optic nerves, similar to human ADOA. 19 Due to decreased mitochondrial fusion activity, disruption of Opa1 GTPase activity results in increased mitochondrial fragmentation, possibly making the retinal ganglion cells more susceptible to apoptosis.20,21 However, further analyses are needed to determine whether mitochondrially-mediated apoptotic pathways are implicated in the pathogenesis of ADOA.
Increased mitochondrial fragmentation due to excessive fission is found in many neurodegenerative disorders, including AD, PD and HD, as well as in ischemic brain damage.5,22
Key players of the fission machinery are Drp1 and Fis1. Drp1 consists of an N-terminal GTPase domain, followed by a helical domain (also termed “dynamin-like middle domain”), an insert B domain, and a C-terminal GTPase effector domain (GED) (Fig. 2). The helical domain allows Drp1 to form ring-like oligomeric structures at the outer membrane of mitochondria to induce mitochondrial fission upon hydrolysis of GTP.5,23,24 A recent case study reported a rare dominant negative mutation in the human Drp1 gene.
11
This mutation inhibits Drp1 oligomerization and thus results in the appearance of elongated mitochondrial morphologies. These abnormal mitochondria lose their respiratory function and can no longer be effectively distributed to regions of high-energy demand, such as neuronal synapses.
25
The dominant negative Drp1 that results from this mutation caused impaired brain development, microcephaly, optic atrophy, lactic academia, and eventual death by 37 days of age.
11
Schematic of Drp1 domain structure and posttranslational modifications. Drp1 has a GTPase domain, a middle domain, an insert B domain, and a GTPase effector domain (GED). Location of posttranslational modifications (PTMs) are indicated by P (phosphorylation), N (S-nitrosylation), S (SUMOylation), or U (ubiquitination). The effect of PTMs on mitochondrial fission activity is indicated in green (activating) or red (inactivating).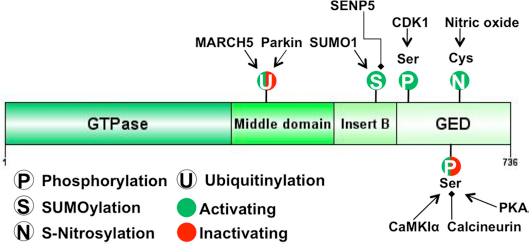
Additionally, Drp1 activity can be tightly regulated by a series of PTMs including S-nitrosylation, phosphorylation, SUMOylation and ubiquitination, as depicted in Figure 2. These PTMs generally do not target the Drp1 GTPase domain but instead are located within or in close proximity to the GED domain, thereby allosterically influencing GTPase activity. 22
PTM Regulation of Drp1 Activity and Mitochondrial Dynamics
Recently, we discovered that Drp1 is a target of S-nitrosylation, a covalent modification of free thiol by a nitric oxide (NO) group, forming an S-nitrosothiol (R-SNO). Specifically, we found that S-nitrosylation of Cys644 within the GED domain of Drp1 enhances its GTPase activity and results in excessive mitochondrial fragmentation. 12 S-Nitrosylation of Drp1 and its involvement in neurologic disorders will be discussed in detail in later sections.
In addition to S-nitrosylation, Drp1 undergoes multiple other PTMs including phosphorylation, SUMOylation, and ubiquitination. Among these Drp1 PTMs, the effects of phosphorylation are perhaps best studied. For example, it is known that phosphorylation of Drp1 at Ser616 by CDK1/cyclin B can increase its GTPase activity. 26 Consequently, Cdk1-dependent phosphorylation of Drp1 results in an increase in mitochondrial fragmentation. 26 In addition, cyclic AMP-dependent protein kinase (PKA) phosphorylates Drp1 at Ser637 and decreases Drp1 activity by inhibiting molecular interactions. 27 PKA-mediated phosphorylation can be counteracted by calcineurin-induced dephosphorylation of Ser637, resulting in translocation of Drp1 to the mitochondrial membrane.27,28 In an apparently paradoxical manner, however, Calcium/calmodulin-dependent PKA 1α (CamKIα) can phosphorylate the same cysteine (Ser637) as PKA but rather induces recruitment of Drp1 to the mitochondrial membrane to form a complex with Fis1, resulting in increased mitochondrial fission. 29 Therefore, additional studies are needed to clarify the basis for the differential effects of PKA and CamKIα.
Drp1 activity is also be affected by SUMOylation, which occurs within the insert B domain (Fig. 2). The attachment of a small ubiquitin-related modifier 1 (SUMO1) to the lysine residues of Drp1 stabilizes the protein and thus increases mitochondrial fragmentation. 30 Several proteins, including SUMO1, Ubc9, and MAPL, have been shown to mediate Drp1 SUMOylation.30,31 This stabilizing modification can be reversed by sentrin/SUMO-specific protease (SENP5).32,33 In addition to SUMOylation, the lysine residues of Drp1 can be ubiquitinated. Parkin and mitochondrial E3 ligase protein MARCH5 can ubiquitinate Drp1 on lysine residues located in the middle domain. Initial studies reported that MARCH5 activity promoted mitochondrial fusion and elongation, suggesting that MARCH-dependent ubiquitination of Drp1 inhibited its activity.34,35 However, a subsequent report by Karbowski et al. 36 found that MARCH5 activity is required for mitochondrial fission. Although one could speculate that different expression levels of MARCH5 might cause these opposite effects, the physiological action of MARCH5-mediated ubiquitination on Drp1 certainly warrants further investigation.
Additionally, parkin-mediated ubiquitination of Drp1 results in its proteasomal degradation. 37 Parkin is a RING-type ubiquitin E3 ligase expressed in many human tissues, including brain, heart, testis, and skeletal muscle. 38 In the human brain, parkin is primarily expressed in neurons in various regions, including the substantia nigra in the midbrain as well as in the CA1/3 region of the hippocampus. 39 Since mutations in the parkin gene cause a familial form of PD, 38 it is tempting to speculate that abnormal regulation of the parkin-Drp1 pathway, which would lead to impaired mitochondrial dynamics, might contribute to the pathogenesis of PD.
S-Nitrosylation, Enhanced Mitochondrial Fission, and Neurodegeneration
NO/Reactive Nitrogen Species and Neurodegeneration
Over the past twenty years, numerous reports identified NO as a crucial protein-modifying molecule in cell biology, affecting many target proteins through S-nitrosylation.40,41 Depending on the target protein, the effects of S-nitrosylation can be quite diverse. NO is typically generated from NO synthase (NOS) during the conversion of L-arginine to L-citrulline. NO production can be controlled by transcriptional activation of inducible NOS (iNOS), or by Ca2+/calmodulin dependent activation of neuronal or endothelial NOS (nNOS or eNOS).
42
A well-characterized pathway for NO production in the nervous system occurs via activation of the N-methyl-D-aspartate-type glutamate receptor (NMDAR).40,43 NMDAR stimulation results in the influx of Ca2+, which, together with calmodulin, stimulates nNOS to generate NO (Fig. 3).
NO signaling pathways in the nervous system. Left: Under physiological conditions, basal levels of NO from nNOS, stimulated by normal synaptic activity of N-methyl-D-aspartate-type glutamate receptors (NMDARs), provide neuroprotection via sGC/cGMP pathways as well as S-nitrosylation of NMDARs and caspases. Right: Under pathophysiological conditions, overactivation of NMDARs, especially extrasynaptic NMDARs, results in increased production of NO. Additionally, iNOS can generate toxic amounts of NO. Excessive NO can lead to S-nitrosylation of Drp1 (forming SNO-Drp1), which contributes to neuronal synaptic injury via excessive mitochondrial fission and bioenergetic impairment. Additionally, S-nitrosylation of PDI, Parkin, XIAP, and GAPDH can contribute to neurodegenerative disorders.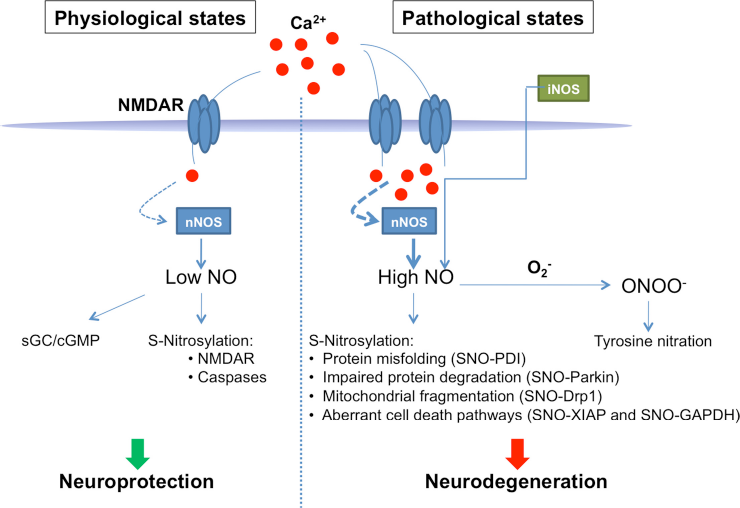
Prior to the discovery of S-nitrosylation, initial studies of NO signaling concentrated on its physiological and neuroprotective functions mediated by activation of soluble guanylate cyclase (sGC) and subsequent production of cGMP (Fig. 3). Subsequently, accumulating evidence has suggested that in a variety of diseases, including AD, HD and PD, nitrosative and oxidative stress appear to trigger aberrant S-nitrosylation events that contribute to neurodegeneration. 40 For example, pathological S-nitrosylation reactions include NO-mediated modifications to parkin and protein disulfide isomerase (PDI) (Fig. 3). 44 46 Additionally, we recently discovered that aberrant S-nitrosylation of Drp1 hyperactivates its mitochondrial fission activity. 12 In this case, NO triggers excessive mitochondrial fission accompanied by increased autophagy (mitophagy) and decreased ATP production, which contributes to synaptic damage conditions such as AD.12,47 In addition to SNO signaling pathways, NO can react with superoxide anion, generated from mitochondrial as well as non-mitochondrial sources (e.g., NADPH oxidase), to form peroxynitrite (ONOO-) (Fig. 3).48,49 Peroxynitrite can result in lipid peroxidation and tyrosine nitration, another NO-mediated PTM that causes pathological alteration in target protein activity, as reviewed elsewhere. 50
S-Nitrosylation of Drp1 and Impaired Mitochondrial Dynamics in AD and HD
AD is the most common neurodegenerative disorder with increasing prevalence because of the aging demographic of our society. AD is clinically characterized by loss of cognitive functions accompanied by intracellular neurofibrillary tangles, composed of hyperphosphorylated tau, and extracellular amyloid plaques, comprised of amyloid-β (Aβ) peptide. Soluble oligomers of Aβ arise from the proteolytic cleavage of amyloid precursor protein (APP) and are thought to contribute to the pathogenesis of AD. Additional evidence accumulated over the past decade has suggested that altered mitochondrial function associated with dysregulated mitochondrial dynamics is another key feature of AD.13,47,51,52
Recently, the neurotoxic Aβ oligomers were reported to cause excessive mitochondrial fragmentation in cell-based models of AD. 12 Specifically, Aβ oligomer-induced generation of NO resulted in S-nitrosylation of Drp1 (forming SNO-Drp1), which was found to hyperactivate its GTPase activity and induce excessive mitochondrial fragmentation. The resulting disruption of mitochondrial bioenergetics caused a drop in ATP levels and consequent synaptic damage. Drp1 as shown to be S-nitrosylated at cysteine residue 644, located in the GED domain. Importantly, mutation of Drp1 Cys644 to an alanine abrogated Ap/NO-induced mitochondrial fission, consistent with the notion that the mitochondrial fragmentation was at least in part mediated by formation of SNO-Drp1. In AD, the only neuropathological correlation with cognitive decline is the degree of synaptic loss. Interestingly, Drp1-C644A (representing a nitrosylation-resistant mutant) prevented Aβ-induced synaptic damage, suggesting that S-nitrosylation of Drp1 at Cys644 contributes to Aβ toxicity in AD. Moreover, the non-nitrosylatable Drp1 mutant prevented nitrosative stress-induced neuronal cell death, lending support to the role of SNO-Drp1 in NO-mediated neuronal damage.
Although this pathway of aberrant SNO-Drp1 formation was initially discovered in human AD brains and animal models of AD, many other neurodegenerative conditions, including PD and HD, are characterized by increased oxidative and nitrosative stress. 53
Consistent with this notion, SNO-Drp1 is present at very early stages in the substantia nigra of PD animal models induced by pesticide exposure. Additionally, SNO-Drp1 appears to contribute to mutant huntingtin (mtHtt)-induced neurotoxicity in cell-based and animal models of HD. 54 HD is an adult onset, genetic neurodegenerative disorder caused by an aberrant expansion of a trinucleotide CAG repeat in the htt gene. The CAG expansion, translated into an expanded polyglutamine (polyQ) repeat in the mtHtt protein, accelerates toxic misfolding of mtHtt, leading to interference with several critical molecular cascades in the cell, and resultant degeneration of synapses and neurons in the affected brain area (i.e., the striatum and cortex).
Similar to the effects of Aβ oligomers on SNO-Drp1, expression of mtHtt significantly increased NO production and subsequently up-regulated the levels of SNO-Drp1 in primary cultures of cortical neurons. Further along these lines, SNO-Drp1 formation was considerably elevated in the striatum of a transgenic mouse model of HD as well as in human postmortem brains from HD patients. 54 In a cell culture model of HD, transfection of a non-nitrosylatable mutant form of Drp1 abrogated the neurotoxic effects of mtHtt on mitochondrial fragmentation and dendritic spine damage, suggesting that SNO-Drp1 is a key mediator of this form of mtHtt toxicity. Taken together, these findings imply that S-nitrosylation of Drp1 may occur in other neurodegenerative conditions, contributing to aberrant mitochondrial dynamics and downstream neuronal damage.
Conclusions and Future Directions
In the present study, we review how PTMs of Drp1, particularly S-nitrosylation, may contribute to excessive mitochondrial fragmentation in neurodegenerative diseases. These findings strengthen the hypothesis that aberrant protein S-nitrosylation may play a key role in the contribution of mitochondrial pathology to many neurodegenerative diseases. Importantly, the demonstration that preventing formation of SNO-Drp1 can ameliorate Aβ- or mtHtt-induced synaptic damage in models of AD and HD, respectively, suggests a new therapeutic approach based on developing drugs to affect disease-related protein S-nitrosylation. Additionally, we anticipate that additional examples of aberrant S-nitrosylation as well as other PTM events will be discovered which influence mitochondrial dynamics and function in a number of other neurodegenerative disorders.
Another important area for future research into the effect of SNO-Drp1 concerns the discovery of endogenous compounds, including nitrosylase and denitrosylase enzymes which regulate its formation and destruction. Related to this question, we recently discovered that SNO-Cdk5 can act as an endogenous nitrosylase, enhancing SNO-Drp1 levels via transnitrosylation of Drp1 (i.e., SNO-Cdk5 donates an NO group to Drp1 to form SNO-Drp1). 55 Further efforts to identify additional molecular controls of aberrant S-nitrosylation of Drp1 will be critical in determining not only the downstream signaling consequences but also for formulating new therapies for several neurological disorders.
Author Contributions
Wrote the first draft of the manuscript: FH and TN. Agree with manuscript results and conclusions: FH, TN, and SAL. Jointly developed the structure and arguments for the paper: FH, TN, and SAL. Made critical revisions and approved final version: FH, TN, and SAL. All authors reviewed and approved of the final manuscript.
Funding
This work was supported in part by grants from the Alzheimer Association (to TN), the Michael J. Fox Foundation (to TN and SAL), and the NIH (P01 ES016738, P01 HD29587, and P30 NS076411) to SAL.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests. Provenance: the authors were invited to submit this paper.