
Abstract
Alteromonas macleodii AltDE1 is a deep sea protobacteria that is distinct from the surface isolates of the same species. This study was designed to elucidate the biological function of amad1_06475, a hypothetical protein of A. macleodii AltDE1. The 70 residues protein sequence showed considerable homology with cold-shock proteins (CSPs) and RNA chaperones from different organisms. Multiple sequence alignment further supported the presence of conserved csp domain on the protein sequence. The three-dimensional structure of the protein was also determined, and verified by PROCHECK, Verify3D, and QMEAN programs. The predicted structure contained five anti-parallel β-strands and RNA-binding motifs, which are characteristic features of prokaryotic CSPs. Finally, the binding of a thymidine-rich oligonucleotide and a single uracil molecule in the active site of the protein further strengthens our prediction about the function of amad1_06475 as a CSP and thereby acting as a RNA chaperone. The binding was performed by molecular docking tools and was compared with similar binding of 3PF5 (PDB) and 2HAX (PDB), major CSPs of Bacillus subtilis and Bacillus caldolyticus, respectively.
Introduction
Extreme environmental conditions such as heat shock, cold shock, and acid shock are destructive for most organisms. For those who survive, adaptation to such stresses is crucial. There are many organisms that have shown these types of adaptive responses, but the actual protein component that is responsible for this adaptation is unknown most of the time. In terms of temperature-related stresses, cold-shock mechanism is much less studied than heat shock one. Although the report of possible existence of cold-shock proteins (CSPs) dates back to the early 1980s, complete elucidation of the cold-shock adapting mechanism has not yet been possible.1,2 With the advanced sequencing methods and more genome-wide information available on databases, bioinformatic analysis on the organisms dwelling low temperature environment might provide helpful information. In this study, we targeted Alteromonas macleodii AltDE1, which are found at around 1000 m below the sea surface. A. macleodii is a common marine microbe under the class γ-proteobacterium. This species is usually separated into two ecotypes; one is the habitant of temperate latitudes and the other is that of deep sea, also known as “deep ecotype” (DE).3,4 The DEs are distinct from the surface isolates and have adapted to the low temperatures of deep sea.
With the recent advancements in sequencing techniques, more and more information regarding genome sequences are available on databases. Consequently, there are increasing amounts of information regarding hypothetical proteins deposited in public sequence database than are experimentally obtained data in Protein Data Bank (PDB).5,6 These proteins are predicted to be expressed from an open reading frame, but do not have any experimental evidence about their functions. 7 Currently, about 50% of the proteins in most of the genome are considered hypothetical protein. 8 This encourages the establishment of computational techniques through the use of experimental data for protein function prediction, including protein interaction networks, phylogenetic profiles, gene expression, sequence alignment data, and homology modelling. 9 Such computational techniques are not sufficient to confirm the biological function of a protein under study, but these results can surely be helpful and work as a guideline for wet laboratory experiments. The sequenced genome of A. macleodii AltDE1 contains approximately 4400 gene sequences, among which more than 1200 are listed as hypothetical proteins. This huge amount of hypothetical proteins might contain valuable information about the cold adaptation mechanism, and exploration of these protein sequences might lead to the discovery of some novel proteins with important practical applications.
Materials and Methods
Sequence retrieval
The NCBI database for A. macleodii AltDE1 10 was initially explored to find out hypothetical proteins with possible interest in research and application. A cold-shock domain containing 70 amino acid residue protein amad1_06475, was selected for the study. The sequence was then stored as a FASTA format sequence for further analysis.
Analysis of physicochemical properties
After retrieving the sequence, we used the ProtParam (http://web.expasy.org/ protparam/) 11 tool of ExPASy for the analysis of the physiological and chemical properties from our protein sequence. This tool can predict different physicochemical properties such as the molecular weight, isoelectric pH, aliphatic index, grand average of hydropathicity (GRAVY), and extinction coefficients.
Homology identification and domain analysis
We used the BLASTp program of NCBI database (http://blast.ncbi. nlm.nih.gov/Blast.cgi) for searching the similarity or homology with our protein against the nonredundant and Swiss-Prot database. For the domain analysis we used the Pfam (http://pfam.sanger.ac.uk/) program (version 27.0) of the Sanger institute. This database is a larger collection of protein families, which is represented by multiple sequence alignments and hidden Markov models. 12 Some motifs were also analyzed by ScanProsite server of Swiss Institute of Bioinformatics (http://prosite.expasy.org/scanprosite/). 13
Structure prediction and model quality assessment
Secondary structure of the protein was predicted by PSIPRED server of UCL Department of Computer Science (http://bioinf.cs.ucl.ac.uk/psipred/), 14 and the three-dimensional structure was predicted by (PS) 2 -v2 server (http://ps2v2.life.nctu.edu. tw/) of Molecular Bioinformatics Center, National Chiao Tung University.15,16 Finally, the quality of the predicted structure was determined by PROCHECK 17 and QMEAN6 18 program of ExPASy server of SWISS-MODEL Workspace, 19 and also with verify3D of the UCLA-DOE (http://nihserver.mbi.ucla.edu/Verify_3D/) Structure Evaluation server. 20
Multiple sequence alignment and phylogenic tree construction
For the sequence conservation identification, multiple sequence alignment was done with Jalview multiple sequence alignment editor (http://www.jalview.org/), and the Phylogenetic tree was constructed by PhyML 3.0: new algorithms, methods, and utilities (http://www.atgc-montpellier.fr/phyml/execution.php). 21
Active site determination
Active site of the target hypothetical protein was determined by metaPocket 2.0 server (http://projects.biotec.tu-dresden.de/metapocket/index.php). This server functions through three steps: calling based methods, meta-pocket site generation, and mapping binding residues. In the initial steps, the provided protein structure is sent to eight predictors, including LIGSITEcs, PASS, Q-SiteFinder, SURFNET, Fpocket, GHECOM, ConCavity, and POCASA, to identify pocket sites on its surface, all the predictors are run at the same time. Finally, it generates pockets (meta-pocket) from all the predictors based on comparative ranking and z-score. 22
Comparative docking study
Docking study was performed by Molegro virtual Docker (MVD) of CLC bio lab. This software runs computations in an integrated environment for studying and predicting how ligands interact with macromolecules. It usually offers high-quality docking depending on a novel optimization technique. 23
Results and Discussion
As the initial approach for the analysis of the target hypothetical protein amad1_06475, the complete protein sequence was analyzed using the ProtParam server, which can predict the physical and chemical parameters for the protein. The molecular weight, isoelectric pH, aliphatic index, extinction co-efficient, instability index, and GRAVY for the target protein sequence according to the ProtParam server output are given in Table S1. These parameters are helpful for experimental handling of the protein, such as for extraction or biological analysis.24,25
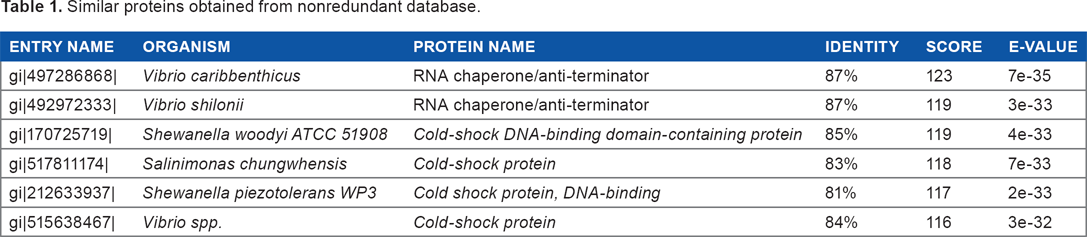
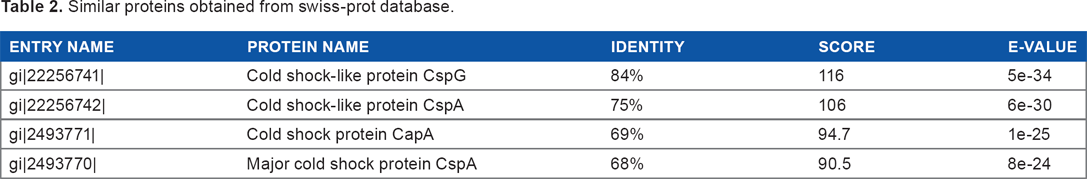
The blastp result against non-redundant and Swiss-Prot database showed homology with CSP and RNA chaperone/anti-terminator (Tables 1 and 2). Pfam server predicted the cold-shock DNA-binding domain at 6–68 amino acid residues with an e-value of 1.1e-30. The cold-shock signature domain was also found in ScanProsite server at 18–37 amino acid residues. Another conserved feature of CSPs is the presence of two nonspecific RNA-binding sequence motifs – ribonucleo-protein motifs 1 and 2 (RNP1 and RNP2, respectively).26,27 The target protein was also found to contain a similar signature sequence at positions 16–23 (RNP1-like motif) and 30–35 (RNP2-like motif) (Figs. S1 and S2).
Similar proteins obtained from nonredundant database.
Similar proteins obtained from swiss-prot database.
The secondary structure of the protein was predicted by the PSIPRED server (Fig. 1) with good confidence of prediction and the tertiary structure of the protein was predicted by the (PS)2-v2 server by using a template 1mjcA with 60.29% identity and 97.14% alignment with the targeted protein. The output of the tertiary structure was found to contain five stranded antiparallel β-barrel sheets (Fig. 2). The existence of such a structure with five antiparallel β-sheets is one of the characteristic features of CSPs. 28 The initial prediction about amad1_06475 as a CSP was also supported by multiple sequence alignment through the conserved alignment of CSP sequences with a common pattern {FGFLxxxxxxxDVFx-HxRxI} (Fig. 3). A phylogenetic tree was also constructed to visualize the evolutionary relationship among different CSPs and our predicted protein (Fig. 4).
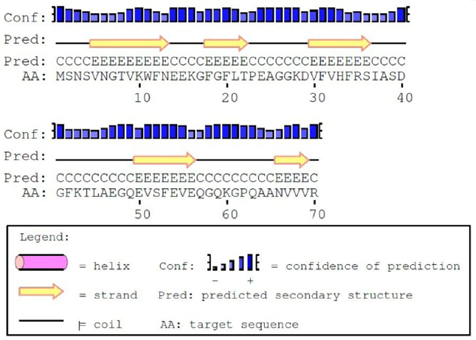
Secondary structure of the protein amad1_06475 predicted by PSIPRED server.
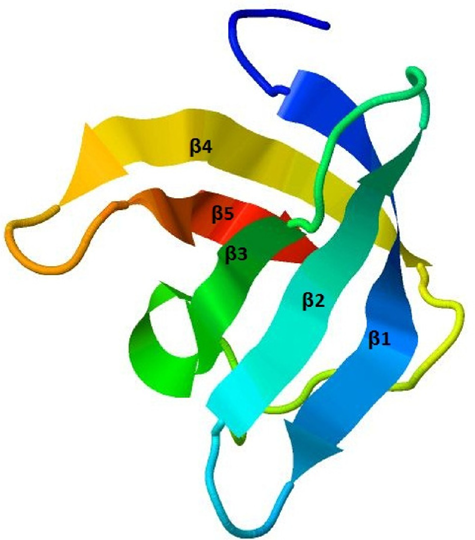
Predicted three-dimensional structure of the protein with five-stranded antiparallel β-barrel sheet.
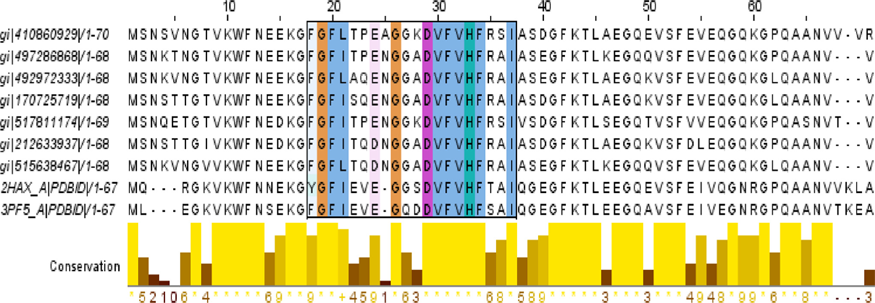
Multiple sequence alignment of different cold-shock proteins (targeted protein at the top row). Bars in the bottom of the figure indicate the conservancy of the cold-shock domain.
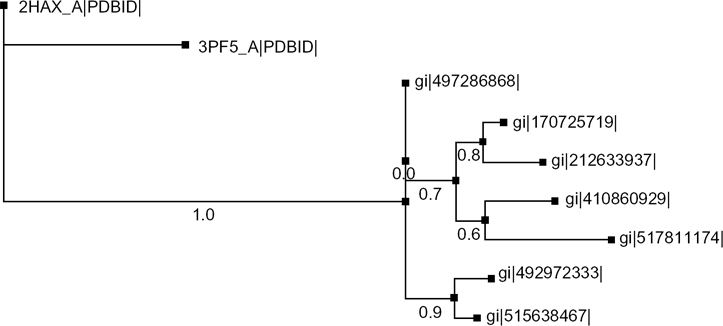
Evolutionary analysis of different cold-shock proteins with the target protein (gi|4108609).
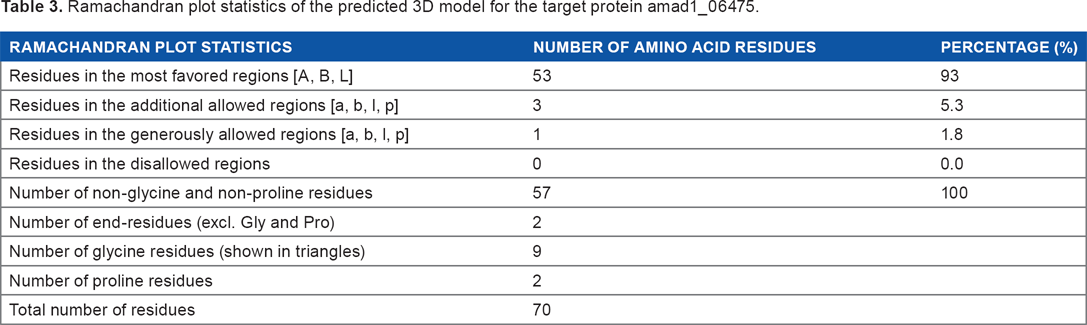
The predicted tertiary structure was validated with the PROCHECK program, which reveals that all the residues are within the limits of the Ramachandran plot (Fig. 5A). The model was presumed to be a good one according to the Ramachandran Plot Statistics, with 93% residues in the most favored regions and no residues in the disallowed regions (Table 3). The QMEAN scoring function estimates the global quality of the models on the basis of a linear combination of six structural descriptors, and four of them are the statistical potentials of the mean force. Here, our protein model was in the dark region of the estimated absolute model quality graph with a global score (QMEAN6 score: 0.764 and Z score: 0.27) (Fig. 5B), which also supported our model quality validation. Finally, the established model of 3D structure for the target sequence was verified by structure validation server (verify3D). The high score of 0.69 in the Verify3D graph indicated that the environment profile of the model was good (Fig. S3).

(A) Ramachandran plot analysis of modeled structure validated by PROCHECK program. (B) Graphical presentation of estimation of absolute quality of model with QMEAN.
Ramachandran plot statistics of the predicted 3D model for the target protein amad1_06475.
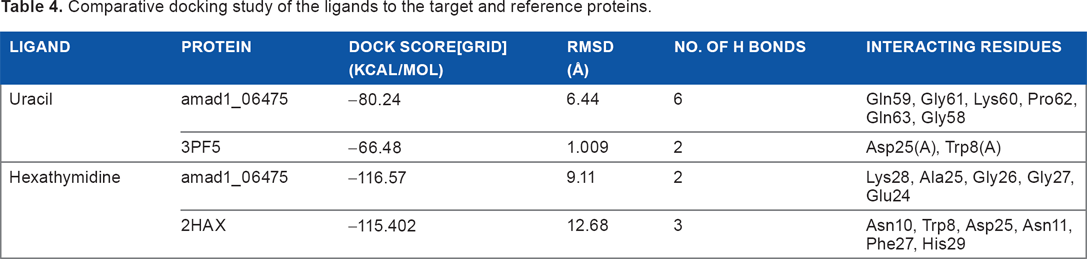
The active site of the protein was predicted by the metaPocket server as shown in Figure 6. From the metaPocket results, we found three pocket/binding sites on the target protein, while two of them were of major concern. The amino acid residues in these two active sites are tabulated in Table S2. For the final confirmation about its role as a CSP, the docking study was performed by MVD. Ligands from various native CSP PDB structures were fetched and docked on the target protein structure. The single uracil molecule from crystal structure of Bacillus subtilis CspB (pdb-3pf5) and hexathymidine from the crystal structure of Bacillus caldolyticus CSP (pdb-2hax) were found to bind amad1_06475 in its active site. For the binding of both the ligands to the target protein, Dock scores were very close to the original bindings of the native CSPs (Table 4). The number of H-bonds and the interacting residues for all these bindings are also compared in Table 4. Binding of both the ligand molecules to the target protein and native CSPs are graphically shown and compared in Figures 7 (uracil) and 8 (hexathymidine). The overall binding of the targeted protein and the two ligands, uracil and hexathymidine, are depicted in Figure 9. CSPs of this kind are thought to bind mRNA like a chaperone and prevent or reduce the formation of mRNA secondary structure to facilitate translation during cold acclimatization.26,27,29 Interactions between ligands and CSPs are divergent among different species. Even within a single species, different CSPs might exhibit different interaction subsites for ligand binding. 30 Hence, the ligands for docking with the target protein were picked from crystal structures of reference CSPs and amad1_06475showed satisfactory docking results in binding to those ligands.
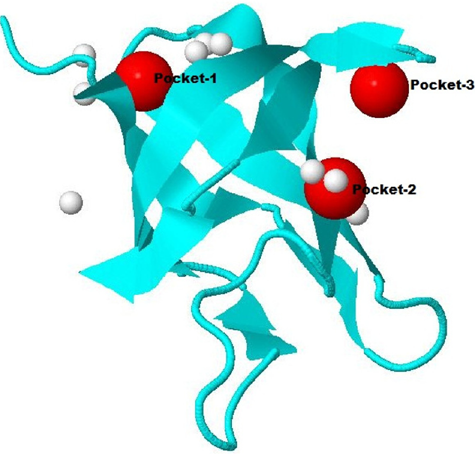
Active sites on the predicted 3D structure of the target protein as determination by metaPocket server (red spheres indicate different meta-pockets).
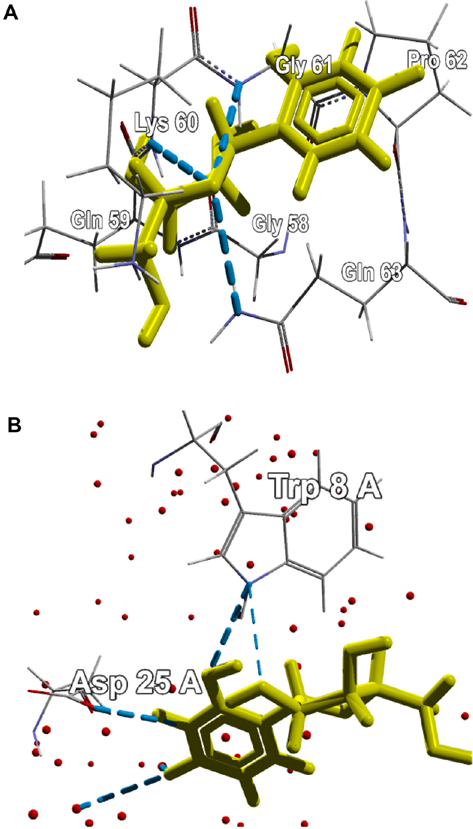
The single uracil molecule docked on (A) the target protein amad1_06475 and (B) 3PF5, a reference cold-shock protein from Bacillus subtilis (blue dashed lines indicate hydrogen bonds and red dots indicate water molecules).

Hexathymidine molecule docked on (A) the target protein amad1_06475 and (B) 2HAX, a reference cold-shock protein from Bacillus caldolyticus (blue dashed-lines indicate hydrogen bonds).
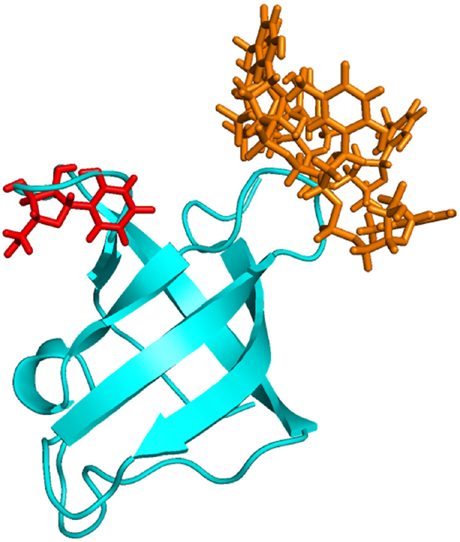
The overall binding between the target protein amad1_06475 and the two ligands, uracil (shown in red) and hexathymidine (shown in orange).
Comparative docking study of the ligands to the target and reference proteins
The NCBI database for A. macleodii AltDE1 currently contains only four proteins (amad1_04180, amad1_10345, amad1_17975, and amad1_18110) with reported functions in cold adaptation or cold responsive biochemical pathways. It is very logical to presume that a habitant of the cold environment of deep sea will contain many more specialized protein molecules to maintain its life and proper functioning at lower temperatures. This study predicts RNA chaperone activity of a CSP that has not yet been reported for this species. With proper experimental validation, this result might help with better understanding of the organism and future direction for research.
Conclusion
Adaptations to stresses are unique properties that are restricted to only the organisms that can withstand the particular stress and actually live with it. The protein components that carry out the adaptation process at the cellular level are of particular interest. These proteins possess huge opportunities as biological tools or to produce new stress tolerant varieties. The present study targeted amad1_06475, a hypothetical protein of A. macleodii AltDE1, and all the analyses suggested the protein to be a CSP with RNA chaperone activity. Even though the target protein was found to contain most of the characteristic features of a CSP, there remains the necessity for experimental validation before the biological function is confirmed. The findings of this article can still be useful for studying other CSPs and finding new ones.
Author Contributions
Conceived and designed the experiments: ARO, SAIA, KMKK. Analyzed the data: ARO, MUH, TPJ. Wrote the first draft of the manuscript: ARO. Contributed to the writing of the manuscript: SAIA. Jointly developed the structure and arguments for the paper: ARO, SAIA. Made critical revisions and approved final version: ARO, SAIA, TPJ. All authors agreed with manuscript results and conclusions, reviewed, and approved the final manuscript.