
Abstract
Microglia are resident mononuclear phagocytes that play a principal role in the maintenance of normal tissue homeostasis in the central nervous system (CNS). Microglia, rapidly activated in response to proinflammatory stimuli, are accumulated in brain lesions of neurodegenerative diseases, such as Alzheimer's disease and Parkinson's disease. The E26 transformation-specific (ETS) family transcription factor PU.1/Spi1 acts as a master regulator of myeloid and lymphoid development. PU.1-deficient mice show a complete loss of microglia, indicating that PU.1 plays a pivotal role in microgliogenesis. However, the comprehensive profile of PU.1/Spi1 target genes in microglia remains unknown. By analyzing a chromatin immunoprecipitation followed by deep sequencing (ChIP-Seq) dataset numbered SRP036026 with the Strand NGS program, we identified 5,264 Spi1 target protein-coding genes in BV2 mouse microglial cells. They included Spi1, Irf8, Runx1, Csf1r, Csf1, Il34, Aif1 (Iba1), Cx3cr1, Trem2, and Tyrobp. By motif analysis, we found that the PU-box consensus sequences were accumulated in the genomic regions surrounding ChIP-Seq peaks. By using pathway analysis tools of bioinformatics, we found that ChIP-Seq-based Spi1 target genes show a significant relationship with diverse pathways essential for normal function of monocytes/macrophages, such as endocytosis, Fc receptor-mediated phagocytosis, and lysosomal degradation. These results suggest that PU.1/Spi1 plays a crucial role in regulation of the genes relevant to specialized functions of microglia. Therefore, aberrant regulation of PU.1 target genes might contribute to the development of neurodegenerative diseases with accumulation of activated microglia.
Introduction
Microglia are resident mononuclear phagocytes that play a principal role in the maintenance of normal tissue homeostasis in the central nervous system (CNS). 1 They are derived from primitive c-kit+ erythromyeloid precursors (EMPs) in the yolk sac emerging as early as on day 8 post-conception during embryogenesis.2,3 EMPs develop into CD45+ c-kitlo CX3CR1– immature A1 cells that subsequently differentiate into CD45+ c-kit– CX3CR1+ A2 cells. Proliferating A2 cells enter into the developing CNS and are incorporated into the brain parenchyma as resident microglia. Microglia have a capacity to constantly scavenge invading pathogens, dying cells, and unwanted synapses by sensing them with a panel of pattern recognition receptors (PRRs). 1 Microglia show a ramified morphology under physiological conditions. When exposed to infectious and traumatic stimuli, they rapidly adopt an amoeboid morphology, followed by secretion of various cytokines, chemokines, and reactive oxygen and nitrogen species. Depending on their microenvironment, microglia are activated to acquire two distinct priming states. Stimulation with lipopolysaccharide (LPS) or interferon-gamma (IFNγ) induces the “classically” activated (M1; proinflammatory) state relevant to defense against bacterial and viral infection, whereas exposure to interleukin (IL)-4 or IL-13 promotes the conversion to the “alternatively” activated (M2; anti-inflammatory) state involved in tissue repair and remodeling. 1
Microglia play a central role in the pathophysiology of human neurodegenerative diseases that are characterized by chronic inflammation associated with accumulation of activated microglia in affected areas, such as Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD).4,5 In AD, amyloid-beta (Aβ) activates microglia by signaling through Toll-like receptors (TLRs) and NOD-like receptors (NLRs), leading to production of proinflammatory mediators potentially toxic to neurons.6,7 In PD, alpha-synuclein (α-Syn), which serves as a danger-associated molecular pattern, directly activates microglia. 8 In HD, mutant huntingtin promotes transcriptional activation of numerous proinflammatory genes in microglia. 9 However, at present, the precise mechanism underlying gene regulation relevant to microglial activation in human neurodegenerative diseases remains largely unknown.
The E26 transformation-specific (ETS) family transcription factor PU.1, also named as Spi1 or Sfpi1 in mouse, acts as a master regulator of myeloid and lymphoid development, expressed chiefly in monocytes/macrophages, neutrophils, mast cells, B cells, and early erythroblasts. 10 PU.1 comprises an N-terminal transactivation domain, a C-terminal DNA-binding domain, and an intervening PEST domain for protein–protein interactions. It activates expression of hundreds of downstream genes by binding to a purine-rich DNA sequence named the PU-box located on the targets. The expression levels of PU.1 target genes are highly variable in different cell types, owing to the difference in cellular concentration of PU.1, chromatin accessibility, motif-binding affinity, and cooperation with neighboring transcription factors. 11 Importantly, PU.1-deficient mice show a complete loss of microglia, along with a lack of mature macrophages, monocytes, neutrophils, and B cells, indicating that PU.1 regulates key genes involved in differentiation and maturation of not only hematopoietic cells but also brain microglia.12,13 However, at present, the comprehensive profile of PU.1 target genes involved in microgliogenesis remains uncharacterized. In the adult human microglia, MCSF (CSF1) treatment elevates the expression levels of PU.1 and stimulates phagocytosis of Aβ, while knockdown of PU.1 reduces their viability and phagocytic capability.14,15 Interferon regulatory factor 8 (Irf8) serves as an essential regulator of development of A2 microglial progenitor cells. 3 Irf8-deficient microglia show fewer elaborated processes with decreased expression of Iba1 and reduced proliferative and phagocytic activities. 16 Runt-related transcription factor 1 (Runx1), whose expression levels are elevated in amoeboid microglia, promotes reverse transition from amoeboid to ramified microglia. 17
Recently, the rapid progress in the next-generation sequencing (NGS) technology has revolutionized the field of genome research. Chromatin immunoprecipitation followed by deep sequencing (ChIP-Seq) serves as one of NGS applications for genome-wide profiling of DNA-binding proteins, histone modifications, and nucleosomes. 18 ChIP-Seq, with advantages of higher resolution, less noise, and greater coverage of the genome, compared with microarray-based ChIP-Chip, provides an innovative tool for studying gene regulatory networks on the whole genome scale. Furthermore, recent advances in systems biology help us to investigate the cell-wide map of the complex molecular interactions by using the literature-based knowledgebase of molecular pathways. 19 Therefore, the integration of high dimensional ChIP-Seq NGS data with underlying molecular networks represents a rational approach to characterize the genome-wide network-based molecular mechanisms of gene regulation. To clarify the biological role of PU.1 in regulation of microglial functions, we attempted to characterize the comprehensive set of ChIP-Seq-based PU.1/Spi1 target genes in microglial cells by analyzing a dataset retrieved from public database.
Methods
ChIP-Seq dataset of microglial cells
A ChIP-Seq dataset of microglial cells was retrieved from DDBJ Sequence Read Archive (DRA) under the accession number SRP036026. The researchers in Dr. Christopher K. Glass's Laboratory, University of California, San Diego, performed the original experiment to study the role of reactive microglia in HD. 9 The raw data are open to public from March 2, 20l4. Currently, no alternative datasets are publicly available for PU.1 ChIP-Seq of microglia. They cloned the N-terminus of wild-type (15Q) or mutant (128Q) human huntingtin in the pCDH-CMV-MCS-EF1-Puro vector (System Bioscience). Either the cloned vector or the empty vector was expressed in BV2 mouse microglial cells, 20 by using the Lentiviral expression system (System Bioscience). Then, they were processed for ChIP-Seq analysis. We studied ChIP-Seq data derived from the cells transduced with the empty vector (no exogenous huntingtin). Following fixation with formaldehyde, sonicated nuclear lysates were immunoprecipitated with a rabbit polyclonal anti-PU.1 (Spi1) antibody (sc-352; Santa Cruz Biotechnology) (SRX451619) or a rabbit polyclonal anti-CCAAT-enhancer-binding protein alpha (C/EBPα, Cebpa) antibody (sc-61; Santa Cruz Biotechnology) (SRX451622). NGS libraries constructed from adapter-ligated ChIP DNA fragments were processed for deep sequencing on Genome Analyzer IIx (Illumina).
First, we evaluated the quality of NGS short reads by searching them on the FastQC program (www.bioinformatics.babraham.ac.uk/projects/fastqc). Then, we removed the reads of insufficient quality by filtering them out with the FASTX-toolkit (hannonlab.cshl.edu/fastx_toolkit). After cleaning the data, we mapped them on the mouse genome reference sequence version mm9 by a mapping tool named COBWeb of the Strand NGS2.0 program, formerly named Avadis NGS (Strand Genomics), or by the Bowtie2 version 2.1.0 program (bowtie-bio.sourceforge.net/bowtie2/index.shtml). Then, we identified the peaks of binding sites with fold enrichment (FE) ≥5 by using the Model-based Analysis of ChIP-Seq (MACS) program or the Probabilistic Inference for ChIP-Seq (PICS) program.21,22 We determined the genes corresponding to the peaks by a neighboring gene analysis tool of Strand NGS in the setting within a distance of 5,000 bp from peaks to genes. We characterized the genomic location of binding peaks by a peak-finding tool of Strand NGS that classifies the locations into the upstream region, 5’ untranslated region (5'UTR), exon, intron, and 3'UTR. We also imported the processed data into a genome viewer named GenomeJack v1.4 (Mitsubishi Space Software). We identified the consensus motif sequences in the genomic regions surrounding the peaks by using the GADEM program. 23
Molecular network analysis
To identify molecular networks biologically relevant to ChIP-Seq-based Spi1 target genes, we imported the corresponding Entrez Gene IDs into the Functional Annotation tool of Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.7 (david. abcc.ncifcrf.gov). 24 DAVID identifies relevant pathways constructed by Kyoto Encyclopedia of Genes and Genomes (KEGG), composed of the genes enriched in the given set, followed by statistical evaluation by a modified Fisher's exact test corrected by Bonferroni multiple comparison test. KEGG (www.kegg.jp) is a publicly accessible knowledgebase that covers a wide range of pathway maps on metabolic, genetic, environmental, and cellular processes, and human diseases, currently composed of 332,680 pathways generated from 466 reference pathways. 25
We also imported Entrez Gene IDs into Ingenuity Pathways Analysis (IPA) (Ingenuity Systems; www.ingenuity.com). IPA is a commercial knowledgebase that contains approximately 3,000,000 biological and chemical interactions and functional annotations with definite scientific evidence. Upon uploading the list of Gene IDs, the network-generation algorithm identifies focused genes integrated in global molecular pathways and networks. IPA calculates the score P-value that reflects the statistical significance of association between the genes and the pathways or networks by the Fisher's exact test.
KeyMolnet (KM Data; www.km-data.jp), a different commercial knowledgebase, contains manually curated content on 164,000 relationships among human genes and proteins, small molecules, diseases, pathways, and drugs. 26 They include the core content collected from selected review articles with the highest reliability. Upon importing the list of Gene IDs, KeyMolnet automatically provides corresponding molecules as nodes on the network. The neighboring network-search algorithm selected one or more molecules as starting points to generate the network of all kinds of molecular interactions around starting molecules, including direct activation/inactivation, transcriptional activation/repression, and the complex formation within one path from starting points. The generated network was compared side by side with 501 human canonical pathways of the KeyMolnet library. The algorithm counting the number of overlapping molecular relations between the extracted network and the canonical pathway makes it possible to identify the canonical pathway showing the most significant contribution to the extracted network.
Results
Identification of 5,264 ChIP-Seq-based Spi1 target genes in mouse microglia
First, we evaluated the quality of ChIP-Seq NGS data examined in the present study. After cleaning, the quality scores mostly exceeded 30 across the bases on FastQC, indicating an acceptable quality for downstream analysis (Supplementary Fig. 1, panels a, b). After mapping them on mm9 by COBWeb, we identified 56,278 Spi1-ChIP peaks detected by MACS and 15,141 Spi1-ChIP peaks detected by PICS. From these, we selected peaks located within a distance of 5,000 bp from protein coding genes, and then extracted 5,264 genes overlapping between data derived from two distinct peak-finding algorithms MACS and PICS termed as the most reliable Spi1 targets (Supplementary Table 1). The peaks were accumulated in the upstream (18.7%) and intronic (69.5%) regions. The motif analysis by GADEM revealed an existence of the PU-box consensus sequences defined as 5‘-GAGGAA-3’ located within the genomic regions surrounding ChIP-Seq peaks (Fig. 1).
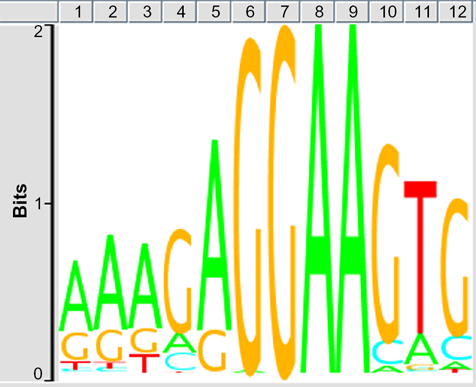
Spi1-binding consensus sequence motif. The consensus motif sequences surrounding Spi1 ChIP-Seq peaks were identified by the GADEM program. The PU-box consensus sequences defined as 5‘-GAGGAA-3’ were located on 80.3% of the peaks detected by MACS.
We identified both Spi1 (FE = 19.3) and Irf8 (FE = 27.8) in the list of Spi1 target genes (Supplementary Table 1; Figs. 2 and 3). Both of them are known to serve as a crucial regulator of differentiation of microglia from EMPs during early embryogenesis. 3 We also found Runx1 (FE = 26.7), a transcription factor acting to constitute a negative feedback loop of PU.1, 27 along with Csf1r (FE = l0.7), Csf1 (FE = 17.8), and Il34 (FE = 31.5), acting as a key growth factor for differentiation of microglia, 28 as Spi1 targets (Supplementary Table 1). Furthermore, we identified known cell type-specific markers for microglia, such as Aif1 (Iba1, FE = 32.9), Cx3cr1 (FE = 17.8), Cd68 (FE = 20.3), Trem2 (FE = 12.8), and Tyrobp (Dap12) (FE = 14.6) in the list of Spi1 target genes (Supplementary Table 1; Supplementary Figs. 2 and 3). Importantly, loss of function of either TREM2 or DAP12, components of a receptor/adapter complex on human microglia, plays a causative role in Nasu–Hakola disease (NHD). 29 Furthermore, we found Syk (FE = 17.8), a downstream signal transducer of the Trem2/Dap12 pathway, as a Spi1 target gene.
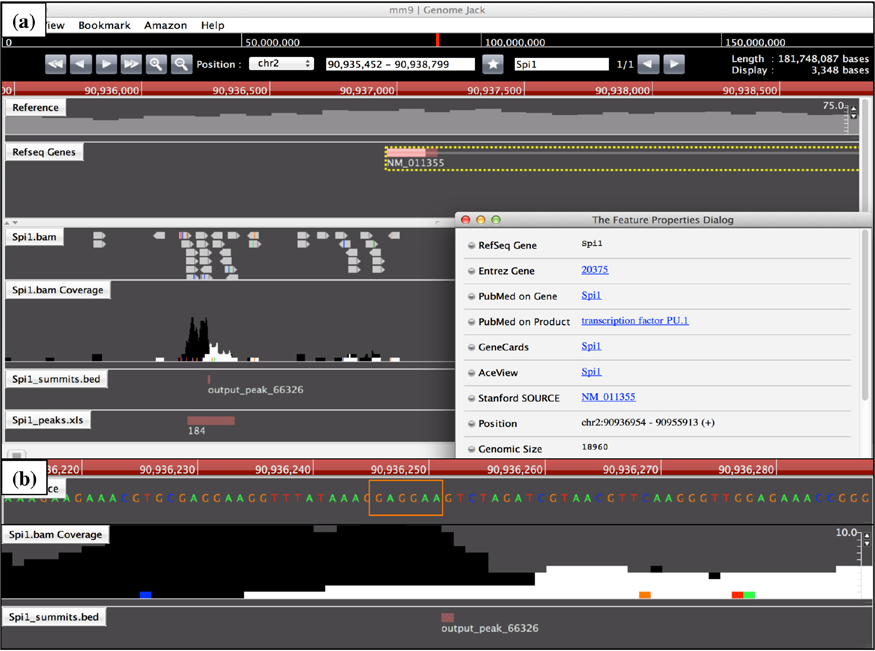
Genomic location of Spi1 ChIP-Seq peak on the Spi1 gene. The genomic location of Spi1 ChIP-Seq peaks was determined by importing the processed data into GenomeJack. An example of transcription factor PU.1 (Spi1; Entrez Gene ID 20375) is shown, where a MACS peak numbered 66326 in the Spi1 .bam Coverage track is located in the promoter region of the Spi1 gene (panel a) with a Spi1-binding consensus sequence motif highlighted by orange square (panel b).
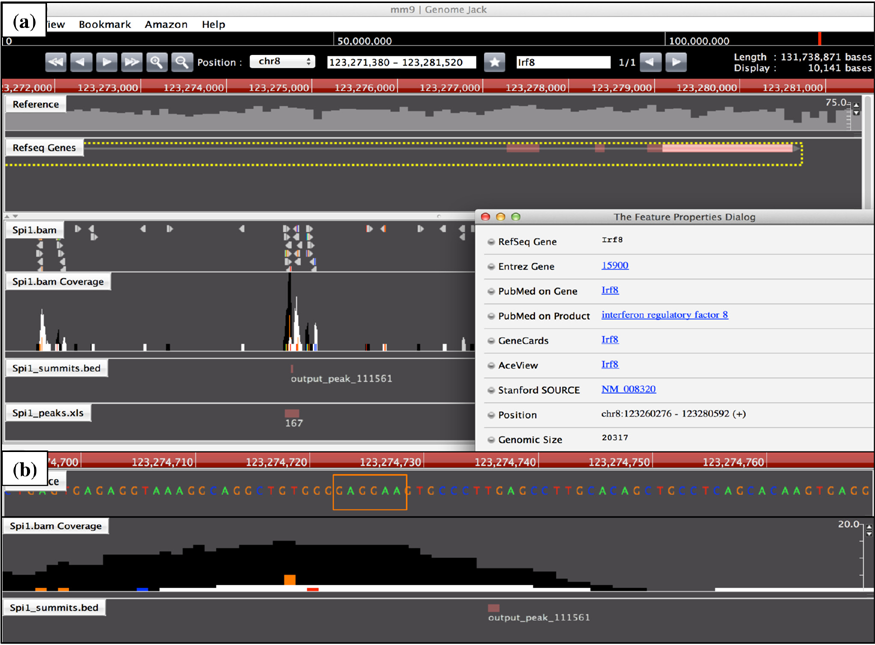
Genomic location of Spi1 ChIP-Seq peak on the Irf8 gene. The genomic location of Spi1 ChIP-Seq peaks was determined by importing the processed data into GenomeJack. An example of interferon regulatory factor 8 (Irf8; Entrez Gene ID 15900) is shown, where a MACS peak numbered 111561 in the Spi1.bam Coverage track is located in the intronic region of the Irf8 gene (panel a) with a Spi1-binding consensus sequence motif highlighted by orange square (panel b).
Next, we studied ChIP-Seq-based Cebpa target genes in BV2 microglial cells. We identified 12,685 Cebpa-ChIP peaks detected by MACS and 10,311 Cebpa-ChIP peaks detected by PICS. From these, we selected peaks located within a distance of 5,000 bp from protein coding genes, and then extracted 3,106 genes overlapping between data derived from MACS and PICS termed as the most reliable Cebpa targets. We found that 1,844 genes are shared between Spi1 targets and Cebpa targets, suggesting the possibility that Cebpa coregulates a substantial proportion (35%) of Spi1 target genes in microglial cells (Supplementary Table 1, underline).
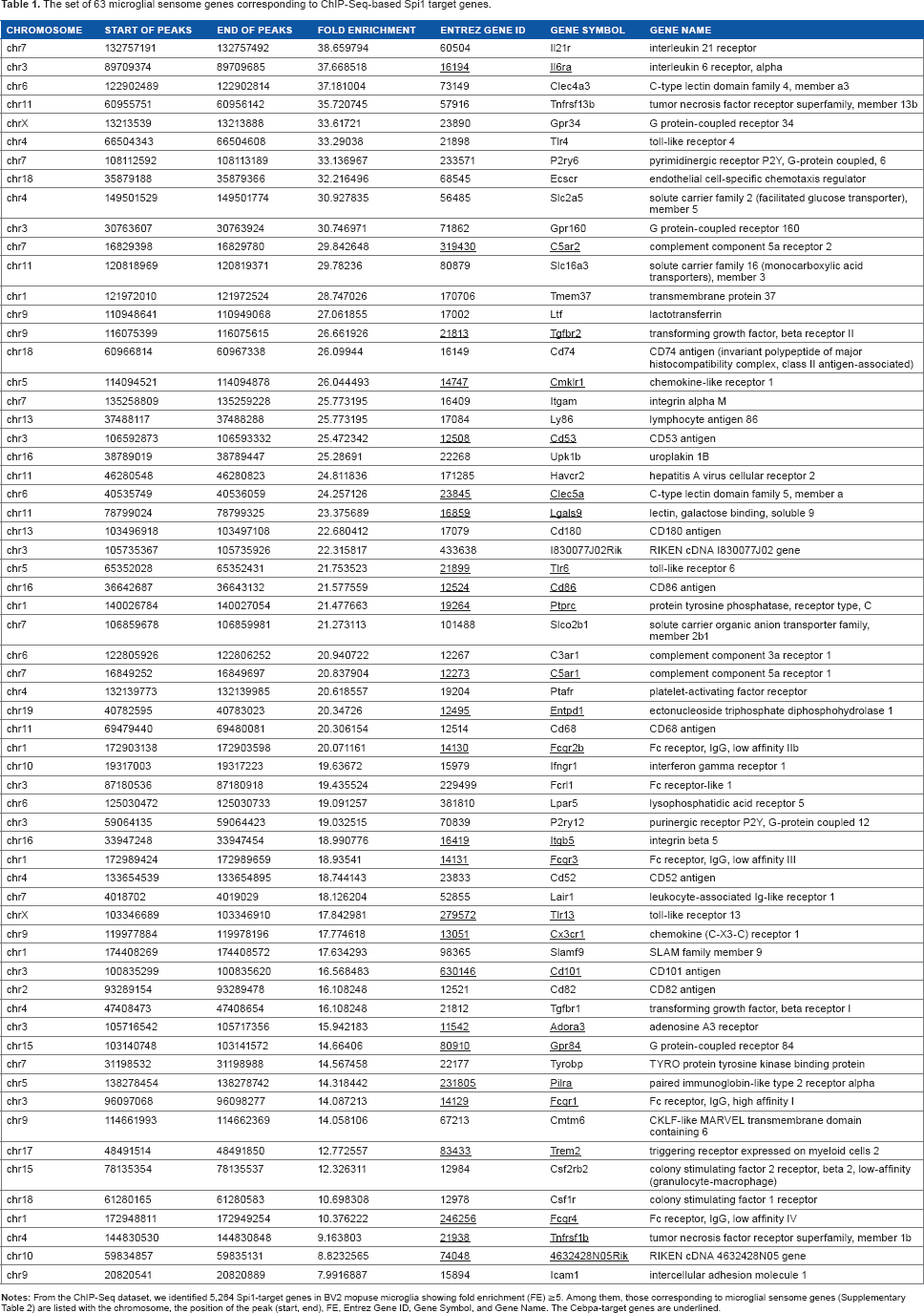
A recent study by direct RNA sequencing of flow cytometry-sorted mouse brain microglia has characterized a set of 100 transcripts exclusively expressed in microglia. 30 The study designated them as “the microglial sensome” (Supplementary Table 2). Importantly, we found that 63 out of 100 microglial sensome genes correspond to ChIP-Seq-based Spi1 target genes, indicating that Spi1 plays a pivotal role in regulation of the genes relevant to specialized functions of microglia (Table 1).
The set of 63 microglial sensome genes corresponding to ChIP-Seq-based Spi1 target genes.
Molecular networks of ChIP-Seq-based Spi1 target genes in microglia
Next, we studied molecular networks of the set of 5,264 ChIP-Seq-based Spi1 target genes by using three distinct pathway analysis tools of bioinformatics. By using DAVID, we identified functionally associated gene ontology (GO) terms. The most significant GO terms included “phosphate metabolic process” (GO:0006796; P = 2.21E-16 corrected by Bonferroni multiple comparison test) for biological process, “plasma membrane” (G0:0005886; P = 1.26E-15) for cellular component, and “GTPase regulator activity” (GO:0030695; P = 1.27E-22) for molecular function.
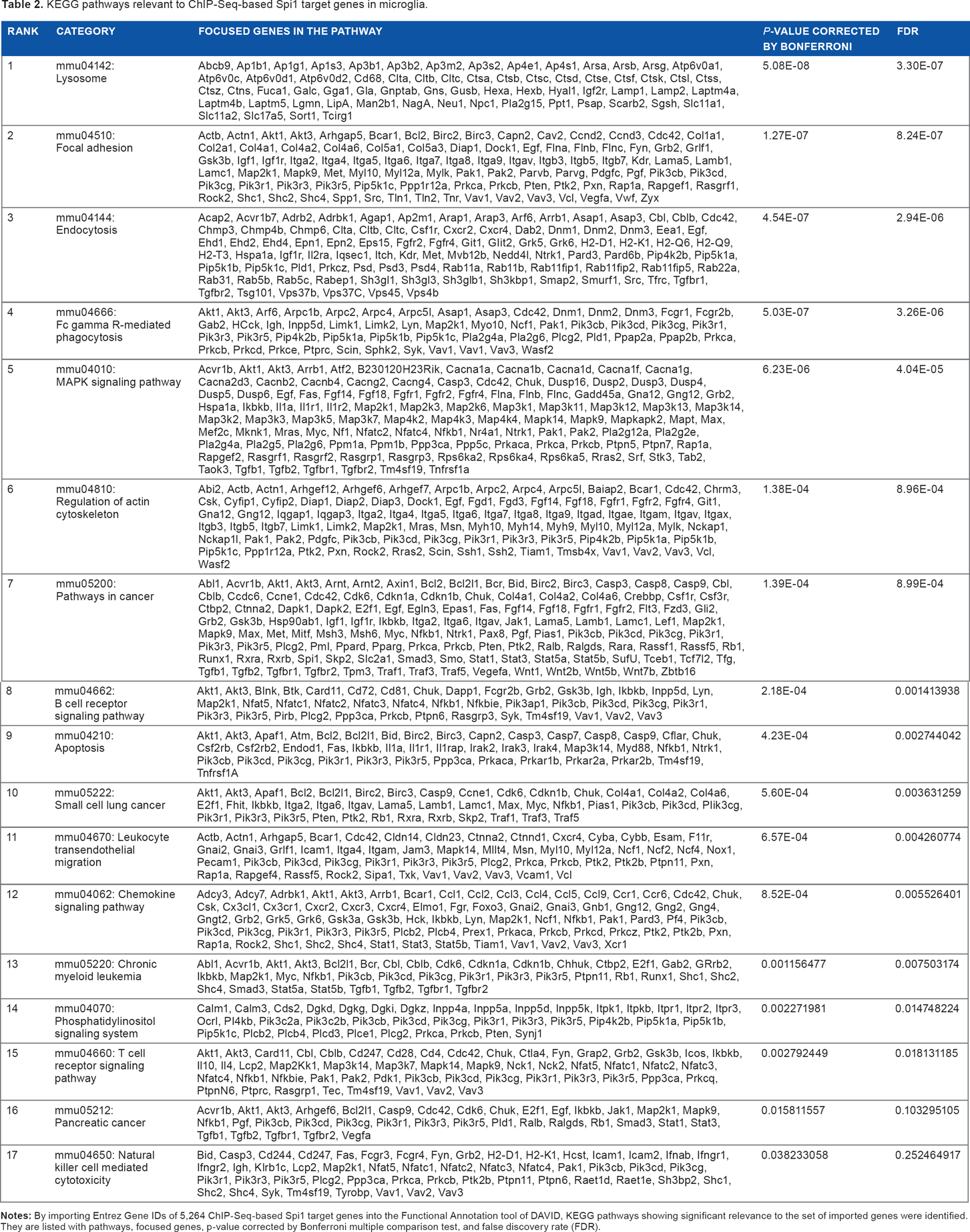
By using KEGG, we found that the set of 5,264 Spi1 targets showed a significant relationship with the pathways defined as “Lysosome” (mmu04142; P = 5.08E-08 corrected by Bonferroni multiple comparison test), “Focal adhesion” (mmu04510; P = 1.27E-07), “Endocytosis” (mmu04144; P = 4.54E-07), “Fcγ receptor-mediated phagocytosis” (mmu04666; P = 5.03E-07) (Fig. 4), and “MAPK signaling pathway” (mmu04010; P = 6.23E-06) (Table 2). Furthermore, they also exhibited significant association with the pathways defined as “Pathways in cancer” (mmu05200; P = 1.39E-04), “B cell receptor signaling pathway” (mmu04662; P = 2.18E-04), “Apoptosis” (mmu04210; P = 4.23E-04), “Leukocyte transendothelial migration” (mmu04670; P = 6.57E-04), “Chemokine signaling pathway” (mmu04062; P = 8.52E-04), and “Chronic myeloid leukemia” (mmu05220; P = 1.16E-03) (Table 2). Importantly, the top-ranked “Lysosome” pathway included the set of 10 cathepsin genes, such as Ctsa, Ctsb, Ctsc, Ctsd, Ctse, Ctsf Ctsk, Ctsl, Ctss, and Ctsz, essential for degradation of lysosomal proteins in microglia (Table 2).
KEGG pathways relevant to ChIP-Seq-based Spi1 target genes in microglia.
Next, we studied molecular networks of 5,264 Spi1 target genes by using the core analysis tool of IPA. They showed a significant relationship with canonical pathways defined as “Fcγ receptor-mediated phagocytosis in macrophages and monocytes” (P = 2.11E-15), “Molecular mechanisms of cancer” (P = 6.92E-15), “B cell receptor signaling” (P = 2.77E-14), “Role of NFAT in regulation of the immune response” (P = 1.17E-12), and “PI3K signaling in B lymphocytes” (P = 2.15E-12). The results of KEGG and IPA combined together indicated that Spi1 regulates expression of not only the genes crucial for normal function of monocytes/macrophages and B cells but also those involved in oncogenesis, particularly in leukemogenesis. IPA also identified functional networks relevant to Spi1 target genes (Supplementary Table 3). The most significant network was defined as “Cell Morphology, Cellular Function and Maintenance, Cell Death and Survival” (P = 1.00E-53), where key components of autophagosomes, such as ATG3, ATG5, ATG7, and ATG10, are clustered (Fig. 5). The second rank network represented “RNA Post-Transcriptional Modification, Cellular Assembly and Organization, Infectious Disease” (P = 1.00E-53).
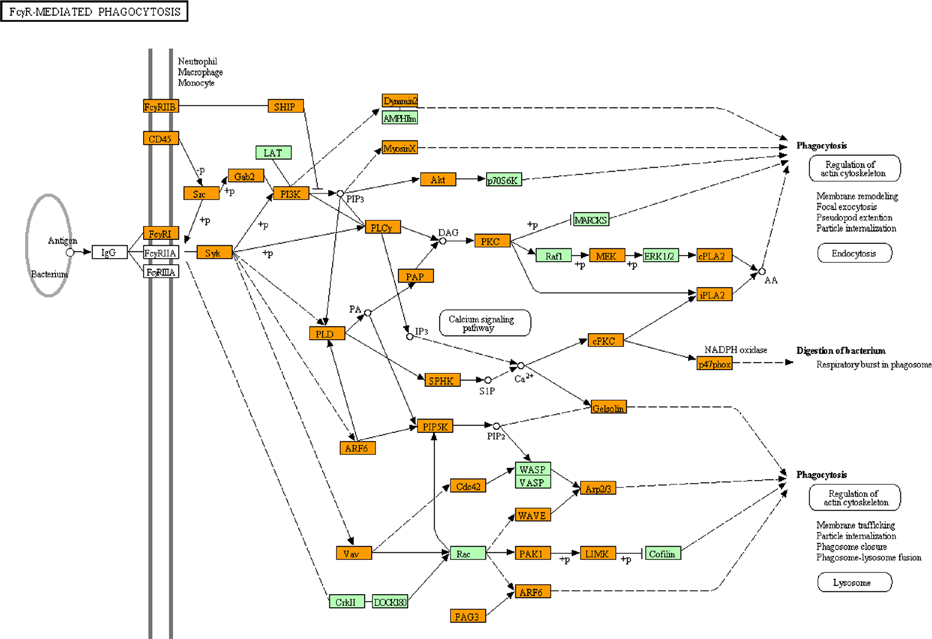
KEGG “Fcγ receptor-mediated phagocytosis” pathway relevant to Spi1 target genes. Entrez Gene IDs of 5,264 ChIP-Seq-based Spi1 target genes were imported into the Functional Annotation tool of DAVID. It extracted the KEGG “Fcγ receptor-mediated phagocytosis” pathway (mmu04666) as the fourth rank significant pathway as listed in Table 2. Spi1 target genes are colored by orange.

IPA “Cell Morphology, Cellular Function and Maintenance, Cell Death and Survival” network relevant to Spi1 target genes. Entrez Gene IDs of 5,264 ChIP-Seq-based Spi1 target genes were imported into the Core Analysis tool of IPA. It extracted the “Cell Morphology, Cellular Function and Maintenance, Cell Death and Survival” network as the first rank significant functional network as listed in Supplementary Table 3. Spi1 target genes are colored by red.
Finally, we studied molecular networks of 5,264 Spi1 target genes by using KeyMolnet. The neighboring network-search algorithm extracted the highly complex network composed of 5,788 molecules and 13,719 molecular relations (Supplementary Fig. 4). It showed the most significant relationship with “Transcriptional regulation by RB/E2F” (P = 7.27E-193). We identified Rb1 (FE = 17.7), Rbl1 (FE = 25.8), Rbl2 (FE = 37.1), and E2f1 (FE = 18.6) as a group of Spi1 target genes (Supplementary Table l).
Discussion
Mice lacking PU.1/Spi1 are devoid of microglia, indicating that PU.1 acts as an indispensable transcription factor for development and differentiation of microglia.13,31,32 By analyzing a ChIP-Seq dataset, we identified 5,264 Spi1 target protein-coding genes in BV2 mouse microglial cells. BV2 cells are derived from immortalized microglia of newborn mouse origin that express morphological, phenotypical, and functional characteristics of primary microglia. 33 We extracted the genes overlapping between data derived from two distinct peak-finding algorithms MACS and PICS termed as the set of most reliable Spi1 targets. Enrichment of the PU-box consensus sequences within the genomic regions surrounding ChIP-Seq peaks validated the reliability of our analysis. By using pathway analysis tools named KEGG and IPA, we found that ChIP-Seq-based Spi1 target genes show a significant relationship with diverse pathways essential for normal function of monocytes/macrophages, such as endocytosis, Fcγ receptor-mediated phagocytosis, and lysosomal degradation, along with the pathway closely related to leukemogenesis. Relevantly, mice with reduced expression of PU.1 develop acute myeloid leukemia. 34 Approximately two-thirds (63%) of “microglial sensome” genes that reflect the microglia-specific gene signature 30 corresponded to Spi1 targets. These observations suggest that Spi1 plays a pivotal role in regulation of the genes relevant to specialized functions of microglia. KeyMolnet constructed the complex network of Spi1 target genes showing the most significant relationship with transcriptional regulation by RB/E2F. PU.1, capable of interacting physically with the C pocket of phosphorylated retinoblastoma (Rb) protein, blocks erythroid differentiation by repressing GATA-1 in mouse erythroleukemia cells. 35
The set of 5,264 Spi1 target genes include Spi1 itself, supporting the previous observations that PU.1 activates its own promoter elements via an autoregulatory loop. 36 We found that approximately one-third of Spi1 target genes in microglia are potentially coregulated by Cebpa, a transcription factor essential for the development of monocytic and granulocytic lineage cells. 37 Importantly, Cebpa directly activates PU.1 gene transcription by binding to its promoter and distal enhancer. 38 A previous study showed that the Cebpa-Spi1 pathway plays a central role in regulation of microglial proliferation in a mouse model of prion diseases. 39
NHD is a rare autosomal recessive disorder characterized by progressive dementia and multifocal bone cysts, caused by genetic mutations of either DAP12 or TREM2. 29 Pathologically, NHD brains exhibit extensive demyelination and gliosis distributed predominantly in the frontal and temporal lobes and the basal ganglia, accompanied by marked accumulation of axonal spheroids and microglia. 40 TREM2 acts as a phagocytic receptor expressed on osteoclasts, dendritic cells, macrophages, and microglia, where it constitutes a signaling complex with an adaptor molecule DAP12, leading to phosphorylation and activation of the downstream kinase Syk. TREM2 expressed on microglia plays a key role in the clearance of damaged neural tissues to resolve damage-induced inflammation. 41 We identified Trem2, Tyrobp (Dap12), and Syk as a group of Spi1 target genes, consistent partly with previous observations. 42 Importantly, Dap12 serves as a hub of the “microglial sensome” network, on which major molecular connections are concentrated. 30 These observations indicate that aberrant function of microglia plays a central role in the pathogenesis of NHD.
A recent study by combining genome-wide linkage analysis and exome sequencing identified several mutations in the CSF1R gene in patients with hereditary diffuse leukoencephalopathy with spheroids (HDLS), a rare autosomal dominant disease that affects predominantly the CNS white matter. 43 Clinically, HDLS exhibits early-onset personality and behavioral disturbances, dementia, and parkinsonism. HDLS shows striking similarities to the pathology of NHD, in view of diffuse demyelination and gliosis with morphologically abnormal microglia and marked accumulation of axonal spheroids, although HDLS never exhibits bone cysts and basal ganglia calcification, both of which are characteristic features of NHD. We identified Csf1r, Csf1, and Il34 as another group of Spi1 target genes, indicating that HDLS represents a disease entity designated as “microgliopathy” caused by microglial dysfunction. Based on these observations, we could propose a hypothesis that microglial dysfunction caused by aberrant regulation of PU.1 target genes contributes to the pathogenesis of various neurodegenerative and neuroinflammatory diseases. Importantly, a recent study indicates that DAP12 acts as a central regulator in gene networks of the late-onset AD. 44
Although ChIP-Seq serves as a highly efficient method for genome-wide profiling of transcription factor-binding sites, the method intrinsically requires several technical considerations to achieve reproducibility of the results. 45 The specificity of antibodies, the sequencing depth and coverage, the source of target cell types and relevant controls, developmental stages, and culture conditions constitute critical factors capable of affecting both genetic and epigenetic features. Motif analysis of a defined set of high-quality peaks makes it possible to evaluate the antibody specificity and to predict the specificity of DNA-protein interaction to some extent. 45 In general, DNA-binding by transcription factors is a highly dynamic process following recruitment of the complex of auxiliary factors, such as coactivators and corepressors. However, in most occasions, ChIP-Seq data generally reflect a snapshot of binding actions of limited DNA-binding factors onto responsive elements, not always corresponding to their biological activities. Because of these limitations, it is highly important to validate main results by examining technical and biological replicates of samples with different sources of ChIP-quality antibodies, along with transcriptome analysis.
Conclusions
By analyzing a ChIP-Seq dataset numbered SRP036026 with the Strand NGS program, we identified 5,264 Spi1 target protein-coding genes in BV2 mouse microglial cells. They included Spi1, Irf8, Runx1, Csf1r, Csf1, Il34, Aif1 (Iba1), Cx3cr1, Trem2, and Tyrobp. Motif analysis identified the PU-box consensus sequences in the genomic regions surrounding ChIP-Seq peaks. By using pathway analysis tools of bioinformatics, we found that ChIP-Seq-based Spi1 target genes show a significant relationship with diverse pathways essential for normal function of monocytes/macrophages, such as endocytosis, Fcγ receptor-mediated phagocytosis, and lysosomal degradation. These results suggest that PU.1/Spi1 plays a pivotal role in regulation of the genes relevant to specialized functions of microglia. Therefore, aberrant regulation of PU.1 target genes might contribute to the development of neurodegenerative diseases with accumulation of activated microglia.
Footnotes
Acknowledgments
The authors thank Ms. Mutsumi Motouri for her invaluable help.
Author contributions
JS designed the methods, analyzed the data, and drafted the manuscript. NA, SK, and YK helped with the data analysis. All authors have read and approved the final manuscript.