
Abstract
We describe a novel computational approach to identify transcription factors (TFs) that are candidate regulators in a human cell type of interest. Our approach involves integrating cell type-specific expression quantitative trait locus (eQTL) data and TF data from chromatin immunoprecipitation-to-tag-sequencing (ChIP-seq) experiments in cell lines. To test the method, we used eQTL data from human monocytes in order to screen for TFs. Using a list of known monocyte-regulating TFs, we tested the hypothesis that the binding sites of cell type-specific TF regulators would be concentrated in the vicinity of monocyte eQTLs. For each of 397 ChIP-seq data sets, we obtained an enrichment ratio for the number of ChIP-seq peaks that are located within monocyte eQTLs. We ranked ChIP-seq data sets according to their statistical significances for eQTL overlap, and from this ranking, we observed that monocyte-regulating TFs are more highly ranked than would be expected by chance. We identified 27 TFs that had significant monocyte enrichment scores and mapped them into a protein interaction network. Our analysis uncovered two novel candidate monocyte-regulating TFs, BCLAF1 and SIN3A. Our approach is an efficient method to identify candidate TFs that can be used for any cell/tissue type for which eQTL data are available.
Keywords
Introduction
Mapping human cell type-specific gene regulatory networks is a significant and longstanding challenge due to the large number (>1,200) of transcription factors (TFs) and the expan-siveness of noncoding regions of the genome. 1 Expression quantitative trait locus (eQTL) studies, in which expression single nucleotide polymorphisms (eSNPs) that are statistically associated with population variation in transcript abundance are systematically mapped, are an increasingly used tool for revealing functional regulatory regions in cell types of interest. 2 The genomic regions that are in linkage disequilibrium (LD) with eSNPs include cell type-specific enhancers (and other types of cis-regulatory elements) that contain TF binding sites (TFBS). However, in order to map cell type-specific regulatory regions to specific TFs, additional information is needed. One approach is to bioinformatically scan–within regulatory regions-genomic sequence for matches to known TFBS sequence patterns3,4; however, this approach has a significant false-positive rate 5 and TFBS sequence preferences may vary depending on the cellular context. 6 Following the recent availability of published binding site locations (based on chromatin immunoprecipitation-to-tag-sequencing [ChIP-seq]) for 133 TFs in various human cell lines through the Encyclopedia of DNA Elements (ENCODE) project, 7 we devised a novel method to directly analyze eQTL regulatory regions using ChIP-seq-based cellular TFBS measurements. Conceptually, our approach builds on previous efforts in which eQTL data were leveraged in motif-based identification of TFBS 8 and in identifying genomic correlates of regulatory SNPs. 9 We applied the method to search for TF regulators using eQTLs from two population studies of human monocytes.10,11 Monocytes are innate immune cells that are vital in host defense, have important roles in many infectious and chronic inflammatory diseases, and are of research interest as models of cellular gene regulatory responses to microbial challenge.12–15
We hypothesized that binding sites of monocyte-regulating TFs are more enriched within monocyte eQTLs than the TFBS that are not monocyte regulators. To investigate this hypothesis, we ranked ENCODE-profiled TFBS ChIP-seq experiments based on the degrees of overlap of their genome-wide binding site profiles (in human cell lines) with monocyte eQTLs. We then tested the enrichment of myeloid cell-derived data sets in the ranked list and the enrichment of known monocyte-regulating TFs in the resulting list of TFs. We found that TFs that are known to have regulatory functions in monocytes are concentrated toward significant
Results and Discussion
To investigate whether anchoring the analysis of TFs using monocyte eQTL data would enable prioritizing TFBS data sets by their relevance to monocytes, we ranked the 397 ChIP-seq-derived TF or cofactor binding site (hereafter, “TFBS”) location data sets based on their overlap (as measured by a z-score of the number of TFBS within the eQTLs; z was computed using a background model based on genome-wide-randomized eQTL placement; “Methods” section) with eQTL regions defined by LD with 80,000 monocyte eSNPs. Then, for two cell types, a cell type related to monocytes (the human myelogenous leukemia cell line K562
16
) and a negative control cell type (the human embryonic stem cell line H1-hESC), we measured the degree to which the data sets of the cell types are enriched at low ranks (corresponding to high overlap with monocyte eQTLs), using the GSEA statistical test.
17
The ChIP-seq data sets from K562 cells were biased toward low ranks (corresponding to high overlap with monocyte eQTLs), with a GSEA score
It is acknowledged that this selection criterion will somewhat bias the
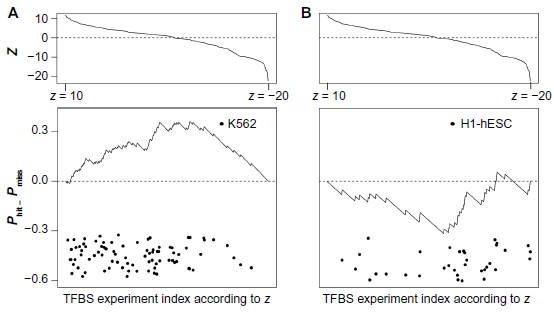
K562 cells are biased toward higher
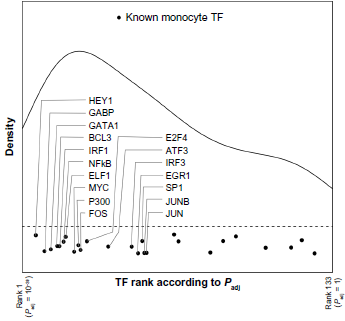
TFs with known functions in regulating monocyte gene expression have higher levels of TFBS overlap with monocyte eQTLs than would be expected by chance. Marks indicate the ranks of TFs with known functions in monocyte gene regulation among all TFs ranked by eQTL overlap significance (
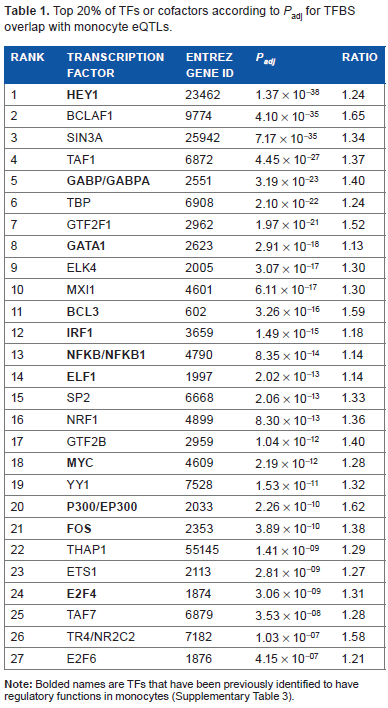
In order to focus on the most probable monocyte-regulating TFs, we selected the top 20% of TFs ranked by
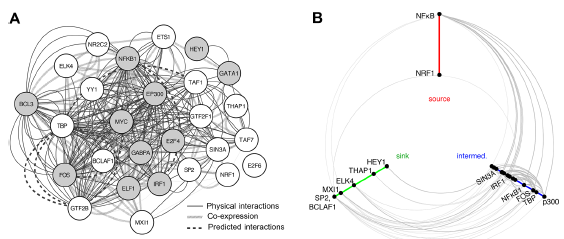
Interaction network of TFs and cofactors identified by overlap analysis with human monocyte eQTLs. (
Top 20% of TFs or cofactors according to
Several of the NMTFs have functions related to immunity or interact with previously identified monocyte-regulating TFs, supporting their potential roles in monocyte gene regulation. For example, the NMTF with the smallest
THAP1, ETS1, TR4, and NRF1 have been previously identified to have specific immune regulatory functions. THAP1 is known to play a role in inducing T-cell apoptosis. 27 STAT3 is a known regulator of ETS1 in inflammation control in mouse macrophages. 28 In addition, a TR4-CD36 pathway is used by mouse miR-133a to regulate lipid accumulation in macrophages. 29 One general transcription factor, TBP whose transcript abundance is thought to be stable in human monocytes, 30 is also in the top 20%; one possible mechanism could be TFIID recruitment to TATA-less promoters by cell type-specific regulators. 31
Discussion and Conclusions
We have demonstrated an efficient method to identify candidate TFs that regulate gene expression in a specific cell type of interest, by leveraging publicly available human genome-wide location data sets and eQTL data sets. A key advantage of this approach is that it can leverage the extensive array of publicly accessible ChIP-seq data from the ENCODE project (1,485 TF ChIP-seq data sets as of the current date). We note that one limitation of the approach is that human ChIP-seq data sets for many TFs are at present primarily available from cancerous cell lines, 7 whose gene regulatory networks may be altered compared to noncancerous cells of the same lineage type. Nevertheless, the strong result for enrichment of known monocyte TF regulators in the list of TFs ranked using our method suggests that this method is useful to screen for TF regulators for noncancerous cell types. Undoubtedly, confirmatory evidence regarding a specific function for a candidate TF in a given cell type of interest, (such as gene expression as we used in this work), would have an important role in avoiding false-positive identification of a TF due to a cancer cell line-specific artifact.
From our analysis using monocyte eQTL data, we conclude that TFs with known functions in regulating gene expression in monocytes tend to have binding sites located in the vicinity of monocyte eQTLs. Of the NMTFs that were identified in our analysis, the two most significant TFs were BCLAF1 and SIN3A. As revealed by protein network reconstruction, the top 20% of TFs identified by our approach are highly interconnected. Further targeted experiments would be useful to elucidate their specific roles in monocyte gene regulation. The approach is generally applicable to other cell types or tissues in that it could be used to identify candidate TFs for any cell type or tissue type for which eQTLs have been mapped from human population studies.
Methods
Software: We used the R statistical computing software (v3.2.2) and Bioconductor (v3.2) packages on OS X 10.11.4.
Databases
We obtained 397 TFBS location data files (derived from ChIP-seq analysis of 133 TFs in 68 human cell lines) from the public repository of the ENCODE project
7
via the University of California Santa Cruz Genome Browser portal. Additionally, we obtained data files containing eSNPs and associated eQTL
eQTL intervals
We mapped genomic intervals containing SNPs that are in LD with each eSNP using the SNP Annotation and Proxy (SNAP) tool
34
using haplotype data from the 1,000 Genomes Project,
35
including only SNPs with
TFBS/eQTL overlap count significance testing
We created a set of allowed regions for random eQTL LD block placement (for use as our background model for TFBS/eQTL overlap counts) by combining peak regions from all 397 TF ChIP-seq data sets with the monocyte eQTLs and merging together any regions whose edges were within 1.9 kbp of one another (selected so that the resulting set of allowed regions covered ~25% of the genome). As a background model for the counts of TFBS/eQTL overlaps, we placed all eQTL LD blocks into random nonoverlapping locations within the set of allowed regions. We carried out 1,000 iterations of the LD block randomization. For each TF ChIP-seq data set, the count of peak center coordinates that overlapped with an eQTL LD block was computed, for both the human eQTL LD blocks and (as a background model) 1,000 iterations of the randomly placed LD blocks. For each ChIP-seq data set, we computed
TF network construction and visualization
We used GeneMANIA
38
(4/16/2016 release) to construct the interaction network for the top 27 TFs (by
Supplementary information
The software source code for this method is available under a free-software, open-source (Apache 2.0) license at github.com/ramseylab/eqtlchiptest.
Author contributions
SAR and MC conceived and designed the experiments. MC and SAR analyzed the data. MC and SAR wrote the manuscript. All authors reviewed and approved the final manuscript.
Footnotes
Acknowledgments
MC thanks Alvin Yu for technical advice. The authors thank Dr Thomas Sharpton for helpful suggestions.
Supplementary Material