
Abstract
Evidence of cardiotoxicity in the preclinical testing of drugs will often lead to compound attrition. The standard method for identifying cardiotoxic compounds involves histopathological analysis of tissue sections, a resource intensive process. In an effort to reduce attrition and capture safety endpoints early within the drug discovery paradigm, a more rapid assessment of target organ effects is desired. Here we describe the results of a preliminary study in which a group of common genes were affected by in vivo exposure to compounds known to cause dose-dependant cardiotoxicity. Adult male Sprague-Dawley rats were treated intraperitoneally with a single dose of digoxin (20 mg/kg), doxorubicin (30 mg/kg), isoproterenol (70 mg/kg), lipopolysaccharide (10 mg/kg) or carbon tetrachloride (800 mg/kg) and euthanized either 6 or 24 hours post-dose. Digoxin, doxorubicin, isoproterenol, and lipopolysaccharide were chosen for this study based on their diverse mechanisms of cardiotoxicity. Carbon tetrachloride, a known liver toxicant, was chosen as a non-cardiotoxic negative control. Genes commonly affected by all four cardiotoxic compounds were grouped together as a list of potential biomarkers. Gene expression changes were subsequently quantified using quantitative PCR. These genes were compared to those affected by novel experimental compounds previously shown to cause cardiotoxicity in rats. These compounds also affected over half of the genes on the biomarker list, whereas the non-cardiotoxic control compound did not affect any genes on the biomarkers list. These data indicate that measuring changes in gene expression could aid in the prioritization of compounds before they are tested in more resource intensive studies.
Introduction
A crucial step in the discovery and development of novel drugs is the conduct of preclinical safety studies conducted prior to clinical trials in humans. Studies to assess safety in the support of regulatory filings are resource intensive, requiring large quantities of chemical matter, animals, and human resource to conduct. Therefore, the safety assessment of new chemical entities needs to occur much earlier in the drug discovery paradigm prior to the commitment of significant resources. An ideal screening strategy for screening of compounds would involve assays that are resource sparing with respect to compound requirements, time, human resources and animal usage. Histological examinations of tissues can take weeks; therefore an alternative endpoint needs to be established to rapidly assess a compounds risk within discovery. Toxicogenomics may provide a rapid assessment of target organ effects after exposure to new chemical entities. These studies will not serve to replace classic endpoints such as histopathology; however they can expedite compound selection by flagging higher risk compounds prior to more resource intensive experiments.
The finding of cardiac toxicity in pre-clinical safety studies often leads to compound attrition, largely due to the relative inability of the myocardium to recover from a significant insult without lasting detrimental effects. The current assessment of cardiac injury in pre-clinical studies is through cardiac histopathology and the use of serum biomarkers. Histopathology is resource intensive and requires assessment by highly trained pathologists. Serum biomarkers such as creatine kinase MB and lactate dehydrogenase are rapid, but not always tissue-specific, and while troponin assays offer the advantage of specificity and sensitivity they likely only detect injury after cardiac cells are damaged such that their cellular membranes are compromised
It has been suggested that activation of genes and signaling pathways is an early response of the heart to toxic insults, and that examination of these genes and pathways will facilitate an understanding of cardiotoxicity (Kang, 2001). In the form of toxicogenomics, gene expression profiling has been proposed as an early sensitive indicator of target organ toxicity (Kramer and Kolaja, 2002; Orphanides, 2003; Shioda, 2004). In this preliminary study, changes in cardiac gene expression were induced with a diverse set of compounds known to elicit cardiotoxicity through a variety of mechanisms. The goal was to identify a set of common genes affected relatively soon after treatment with test compounds, before histological lesions or serum biomarkers would be detected. In this manner, changes in gene expression could serve as early biomarkers for cardiotoxicity, and facilitate the prioritization of compound selection and the design of further preclinical tests.
Digoxin (DIG), doxorubicin (DOX), isoproterenol (ISO), and lipopolysaccharide (LPS) were chosen based on their diverse mechanisms of action and known cardiotoxic potential in rats. Carbon tetrachloride (CCL4), a known liver toxicant in rats, was chosen as a non-cardiotoxic negative control. DOX cardiotoxicity is well studied, with multiple mechanisms of toxicity such as oxidative damage, mitochondrial dysfunction and activation of apoptotic pathways (Childs et al. 2002; Lou et al. 2005; Xu et al. 2001; Zhou et al. 2001). DIG is a cardiac glycoside that is used for the treatment of congestive heart failure, but has the potential of causing cardiac toxicity at higher doses (Fisch and Knoebel, 1985). DIG cardiotoxicity in the rat is not extensively characterized, however acute toxicity studies of analogous cardiac glycosides have been performed (Scott et al. 1971). ISO can induce cardiac myocyte death at high doses (Burniston et al. 2005; Rupp et al. 1994), with a mechanism of action that likely stems from its catecholaminergic affects and subsequent oxidative tissue injury. The bacterial endotoxin LPS, is known to induce sepsis in laboratory animals, with subsequent cardiotoxic effects that can lead to severe and occasionally lethal hypotension (Chagnon et al. 2005 and 2006; Comstock et al. 1998). AG 12917 and AG 12986 are structurally similar cyclin-dependent kinase (CDK) inhibitors, which were developed as potential chemotherapeutics. They were subsequently discontinued from development due to toxic liabilities in preclinical models, including cardiac myopathy in rodents and non-rodents observed in histological sections. The mechanisms of toxicity for these CDK inhibitors is unknown.
Although delayed ventricular repolarization (QT prolongation) is a significant concern for small molecule drugs, affects related to QT prolongation were not evaluated in this study. As stated in the ICH Guideline for the Non-Clinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT interval prolongation) by Human Pharmaceuticals, the ionic mechanisms of repolarization in adult rats and mice differ from larger species, including humans, and therefore, rats are not an accepted model for assessing QT prolongation.
The objective of these studies were to determine if there are common genes affected by cardiotoxic compounds in rats. Genes with robust expression patterns were verified with quantitative polymerase chain reaction methods (Q-PCR). To test the hypothesis that a set of genes could act as early biomarkers for rat cardiotoxicity, these biomarker genes were matched to those affected by pre-clinical compounds with known cardiotoxic effects. Those transcripts that appear predictive of cardiac toxicity in the rat are described.
Materials and Methods
In Vivo
Male Sprague-Dawley rats weighing between 175–220 grams were used for all studies. Animals were purchased from Charles River Laboratories (Kingston, NY) and allowed to acclimate for one-week prior to use. All animals were given food and tap water ad-libitum, and housed under a 12-hour light/dark cycle. Rats were treated in accordance with the Pfizer Global Research and Development Institutional Animal Care and Use Committee guidelines, and with the NIH Guide for the Care and Use of Laboratory Animals (NRC, 1996). Test compounds were given by single intraperitoneal injection in normal saline, with the exception of carbon tetrachloride, which was formulated in corn oil. AG 12986 and AG 12917 were dosed intravenously in 100 mM sodium chloride, 50 mM sodium acetate, pH 4.5.
Dose range-finding studies (data not shown) were completed with time points 6, 24 hours and 7 days after dosing. The doses selected for subsequent gene expression studies approached the maximum tolerated dose but did not cause early overt toxicity. Accordingly, for gene expression studies animals were randomly divided into groups (n = 6/time point) and given a single intraperitoneal injection of either normal saline vehicle (VEH), 20 mg/kg of DIG, 30 mg/kg of DOX, 70 mg/kg of ISO, 10 mg/kg of lipopolysaccharide LPS, 800 or 160 mg/kg of CCl4, 70 mg/kg of AG 12917 (4-{[4-amino-5-(2,6-difluorobenzoyl)-1,3-thiazol-2-yl]amino}-N-{[(2S)-1-met hylpyrrolidin-2-yl]methyl}benzamide hydrochloride) or 60 mg/kg of AG 12986 ({[4-amino-5-(2, 6-difluorobenzoyl)-1, 3-thiazol-2-yl]amino}-N- [(1R)-2- (dimethylamino)-1-methylethyl] benzamide). Six or 24 hours later, animals were euthanized with a lethal intraperitoneal dose of Nembutal. Six hour time points were chosen to capture relatively early changes in gene expression. Samples were collected 24 hours after dosing because this time point is commonly utilized in acute toxicology studies during early drug safety research. Biopsy punches were used to remove approximately 100 mg of ventricular tissue, which were flash frozen in RNA-later (Qiagen, Valencia, CA) for subsequent RNA isolation and gene expression analysis. Another group of animals were euthanized one week after dosing to assess morphological changes in the heart.
RNA preparation and DNA microarray
The RNA isolation procedure followed the Affymetrix GeneChip Expression Analysis technical manual (Affymetrix Inc., Santa Clara, CA). Briefly, total RNA was isolated from the samples using a Qiagen RNAeasy fibrous tissue protocol (Qiagen, Valencia, CA). RNA quantity and quality were assessed by an optical density reading at 260/280 wavelengths and by analysis with agarose gel electrophoresis. Approximately 5 μg of RNA, from four of the six samples displaying the highest quality, were pooled. Some samples yielded low quality RNA, and subsequent pooling of samples was limited to four per group to preserve equal sample size among groups. cDNA synthesis was performed using the Superscript Choice system (Invitrogen Life Technologies, Carlsbad, CA) with an oligo(dT) primer containing a T7 RNA polymerase promoter. Aliquots of RNA were also saved for cDNA synthesis of individual samples. In Vitro transcription was employed to produce biotinlabeled cRNA using the BioArray RNA transcript labeling kit (Enzo Life Sciences, Inc. Farmingdale, NY). Fragmented cRNA was hybridized to the RG U34A GeneChip oligonucleotide array (Affymetrix Inc, Santa Clara, CA) for ∼16 hrs, stained using an automated fluidics station (Affymetrix Inc, Santa Clara, CA), and subsequently scanned by an Agilent GeneArray Scanner (Agilent, Palo Alto, CA). Fragmented cRNA from the negative control group (carbon tetrachloride) was hybridized to the RG 230-2.0 chip.
Quantitative Q-PCR
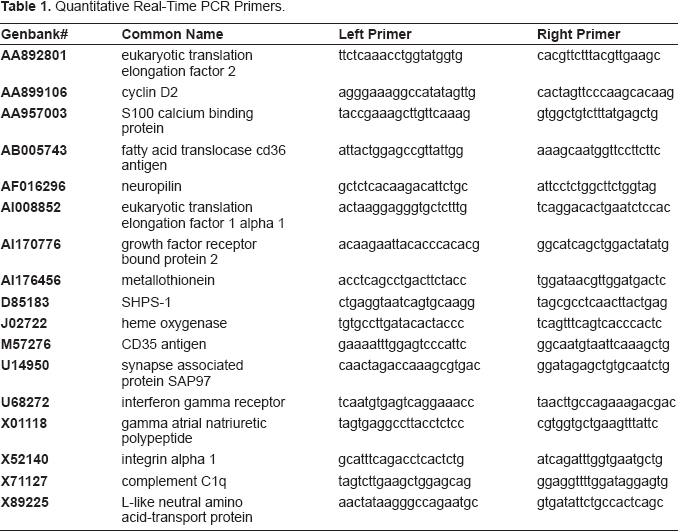
Changes in specific genes of interest were analyzed using quantitative real time PCR (Q-PCR) applied to each individual sample (n = 4), rather than the pooled sample analysis used with the GeneChips. The samples were derived from the same four animals used to pool RNA. This approach provided measurements of variability via statistical analysis. Individual RNA samples were reverse transcribed using a poly dT-oligonucleotide primer to produce single stranded cDNA with Retroscript™ (Ambion Inc., Austin, TX). Primers (Table 1) were selected using on-line software that optimizes primer selection for product size and favorable Q-PCR reaction conditions (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). The target sequence was PCR amplified by combining primer pairs in a solution of PCR Master Mix (Promega, San Luis Obispo CA). PCR conditions were as follows: 30 cycles of denaturation at 94 °C for 30 sec, 45 °C (+/– 5 °C) for 30 sec, 72 °C for 30 sec, 72 °C for 7 min. PCR fragments of expected size (as determined by gel electrophoresis) were cloned using a TOPO TA cloning kit (Invitrogen, Carlsbad, CA) with subsequent plasmid DNA purification using a QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA). Plasmid DNA, linearized with Bgl II, was then used to generate standard curves for subsequent sample analysis on a LightCycler thermocycler (Roche Diagnostics Corp., Indianapolis, IN). Single stranded cDNA or plasmid standards were amplified for quantitation using the QuantitectTM SYBR Green PCR kit (Qiagen, Valencia, CA). Q-PCR conditions for sample analysis involved initial denaturing at 95 °C for 90 sec, followed by 50 cycles of amplification. Each cycle included denaturing at 94 °C for 15 sec, oligonucleotide annealing at 50 °C for 20 sec, product synthesis at 72 °C for 30 sec, and quantitative acquisition at 1–2 °C below the melting temperature of the product.
Quantitative Real-Time PCR Primers.
Data analysis
Affymetrix MicroArray Suite (MAS 5.0) software was used to analyze image data collected from the gene array scanner. Relative expression levels were calculated using GeneSpring (Silicon Genetics, Redwood City, CA.) software, incorporating normalizations to correct for varying signal strengths between chips. The list of genes selected for follow-up by Q-PCR included genes that were induced or suppressed, by all four compounds, at least 2-fold relative to control samples, but not affected by CCL4. We also specified that inclusion be limited to only those genes found to be present (by MAS 5.0 analysis) in treated samples in the case of induced genes, and present in vehicle control samples in the case of suppressed genes. Microsoft Excel, and GraphPad Prism (fold change values and Students t-test) were used to analyze Q-PCR data.
Results
Six and 24 hours after compound administration, rats did not display measurable increases in serum biomarkers of cardiotoxic injury (CK-MB and troponin I) compared to controls, and high variability within groups was observed (Table 2). One week after intraperitoneal administration of the test compounds, compound-induced morphological changes were detected only in the LPS and the CDK inhibitor treated rats (data not shown). Microscopic changes in the LPS-treated animals were subtle and characterized by multifocal myocardial degeneration and necrosis with concomitant inflammatory cell infiltration, primarily by lymphocytes and macrophages. Compound-related microscopic findings in the heart of rats treated with AG compounds were characterized by scattered individual cell necrosis/apoptosis throughout the myocardial interstitium and occasional multifocal to coalescing areas of hemorrhage, cardiomyocyte degeneration and necrosis.
Serum creatine kinase MB and troponin I levels after acute administration (ip) of test compounds (DIG, 20 mg/kg; DOX, 30 mg/kg; LPS, 10 mg/kg; or ISO, 70 mg/kg) to male rats (n = 4/group).
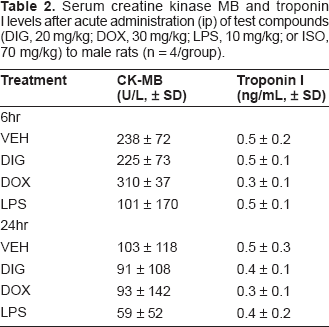
Table 3 shows the genes in common among the cardiotoxicants that were at least 2 fold higher or lower than control levels measured in cardiac tissue 6 or 24 hours after administration using Affymetrix GeneArrays. The induced genes include (Genbank followed by common name): AA892801, eukaryotic translation elongation factor 2; AA957003, S100 calcium binding protein; AI176456, metallothionein; AB005743, fatty acid transporter; AI008852, eukaryotic translation elongation factor 1 alpha 1; AI170776, growth factor receptor bound protein 2; D85183, protein tyrosine phosphatase SHPS 1; J02722, heme oxygenase; M57276, CD53 antigen; U14950, synapse associated protein; U68272, interferon gamma receptor; X89225, l-like neutral amino acid transport protein; and X71127, compliment C1q. All four test compounds suppressed the following genes at least 2 fold lower than control levels measured in cardiac tissue 6 or 24 hours after administration: AA899106, cyclin d2; AF016296, neuropilin; X01118 gamma atrial natriuretic polypeptide; and X52140, integrin alpha 1. These trends of gene induction and suppression obtained from pooled samples were confirmed using quantitative PCR (Fig. 1A–1D) from individual rat samples.
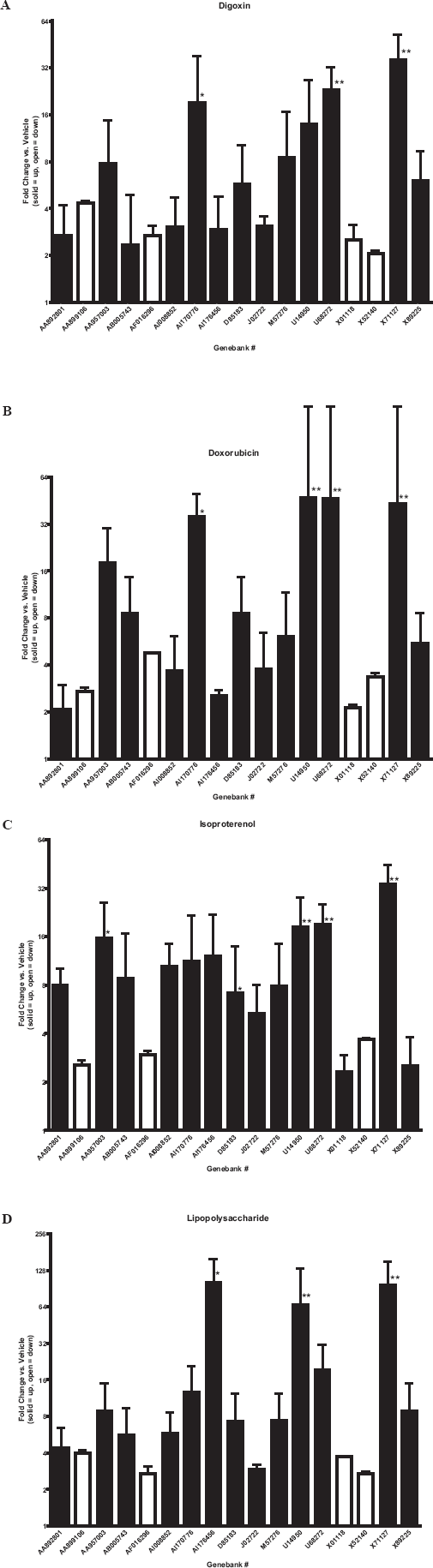
Quantative real-time PCR: gene expression relative to vehicle treated controls. Total RNA extracted from cardiac tissue of male rats (n = 4/group), 6 or 24 hours after acute administration (ip)of test compounds (DIG, 20 mg/kg; DOX, 30 mg/kg; LPS, 10 mg/kg; ISO, 70 mg/kg). Solid bars indicate induction and open bars indicate suppression relative to vehicle treated controls. * = p < 0.05,** = p < 0.01, t-test vs. vehicle treated controls.
Affymetrix GeneChip fold-change values (relative to vehicle treated group) from pooled samples (n = 4/pooled sample). Total RNA extracted from cardiac tissue of male rats, 6 or 24 hours after acute administration (ip) of test compounds (DIG, 20 mg/kg; DOX, 30 mg/kg; LPS, 10 mg/kg; or ISO, 70 mg/kg). ↓ indicates down regulation of gene.
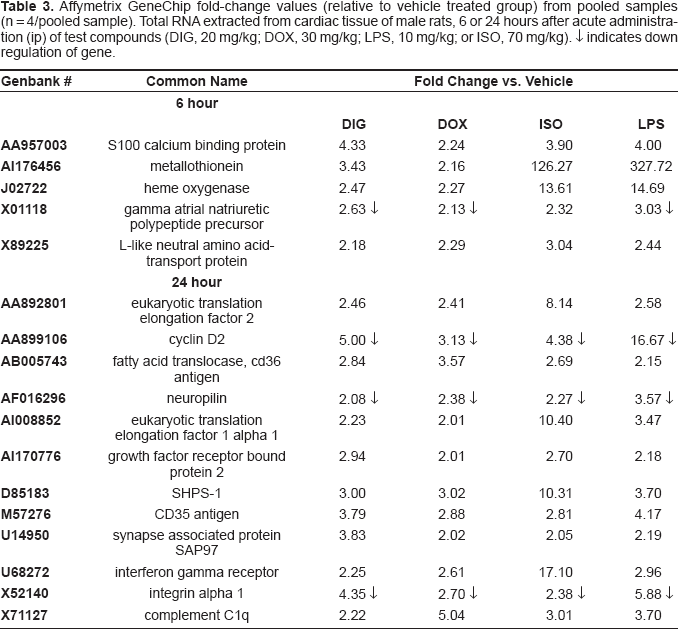
There were 17 genes commonly affected by all four cardiotoxic compounds. None of these genes changed in the cardiac tissue of rats treated with CCL4 (Table 2). Figure 2 depicts a Venn diagram containing these 17 biomarker genes with those affected by AG 12917 and AG 12986. The overlapping regions depict 8 genes that were affected by AG 12917, AG 12986, and all four of the cardiotoxic compounds. Two additional genes affected by either of the AG compounds and all four of the test compounds were observed. Listed by Genbank number followed by common name, these genes were: AA892801, eukaryotic elongation factor 2; AA899106, cyclinD2; AA957003, S100 calcium binding protein; AB005743, fatty acid transporter; AF016296, neuropilin; AI176456, metallothionein; AI170776, growth factor receptor bound protein 2; X52140, integrin alpha-1; X01118, gamma atrial natriuretic polypeptide; and X89225, L-like neutral amino acid transport protein.
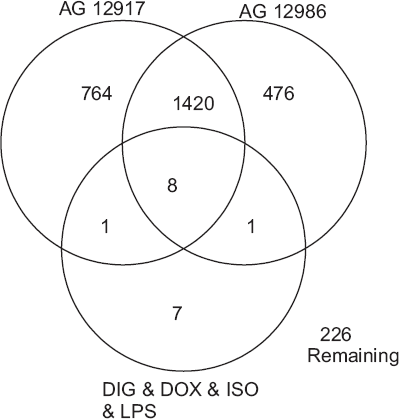
Affymetrix GeneArrays: Venn diagram includes genes affected by test compounds (DIG, 20 mg/kg; DOX 30, mg/kg; LPS, 10 mg/kg; or ISO, 70 mg/kg) or novel cyclin-dependent kinase (CDK) inhibitors (AG 12917, 70 mg/kg; AG 12986, 60 mg/kg). Total RNA extracted from cardiac tissue of male rats (n = 4 pooled samples per chip) 6 or 24 hours after acute administration (ip) of compounds. Overlapping regions indicate commonly affected genes.
Discussion
Several genes on our list were particularly noteworthy, as their involvement in cardiac stress has been noted in scientific literature. Interestingly, the direction of change (induced or suppressed) was uniform across genes with the exception of the gamma atrial natriuretic polypeptide precursor gene, where expression was induced by ISO but suppressed by the other compounds. Literature suggests the natriuretic peptides are involved in cardiac hypertrophy and affected by adrenergic agents such as phenylephrine (Knowlton et al. 1991; Levin et al. 1998; Silberbach et al. 1999), which may explain the up regulation of a component of this pathway after ISO administration. Suppression of the gene by the other three compounds warrants additional study. For this particular gene, the direction of change may relate to compound specific mechanisms. Compounds presumably affect gene expression patterns at different rates and magnitudes, such that a gene may be induced early on, but suppressed at later time points due to tissue damage or negative feedback mechanisms for example. Those analysis are beyond the scope of these studies but futures studies could follow gene expression changes across time and yield important information for potential biomarker discovery.
In vitro and in vivo studies suggest that the D-type cyclins may be crucial regulators of cardiac hypertrophy in response to stressors (Busk et al. 2002). Additionally, a review by Brinkmann and colleagues (Brinkmann et al. 2002) includes studies that suggest fatty acid translocase CD36 proteins may play a role in the etiology of cardiac hypertrophy. Another review by Nath and colleagues (Nath et al. 2000) discussed the role of metallothionein during cardiac oxidative stress. In vivo studies suggest heme oxygenase expression can be affected by ischemic events and that this heat shock protein may be involved in cardiac cellular protection pathways (Lakkisto et al. 2002). Growth factor receptor bound protein 2 may playa role in cardiac hypertrophy and fibrosis (Zhang et al. 2003). Finally, integrins are believed to play a role in healing and remodeling after myocardial injury in rats (Newata et al. 1999). The biochemical pathways and extent of involvement in cardiac stress have yet to be elucidated for many of these genes. Genes that did not readily appear in current literature require further study, but we speculate they are involved in cardiac growth and repair responses to stress.
In this study a small selection of compounds known to cause dose-dependant cardiac toxicity were administered to rats in an effort to measure the subsequent acute changes in heart tissue gene expression and derive a list of potential expression biomarkers of cardiac toxicity. There were 17 genes affected in common to all four test compounds, and these affects occurred within an acute setting without the presence of overt morphological or biochemical changes. Eight of these 17 genes were also affected in rat heart tissue after single dose treatment with investigative compounds AG 12986 and AG 12917, with 1 additional gene affected by the individual AG compounds. AG 12986 and AG 12917 also induced cardiotoxicity in previous studies that led to their attrition in development, but relied on histopathology as the endpoint. It is conceivable that a single dose study using gene expression endpoints could be used as an early and rapid screen by testing a larger cohort of compounds in effort to aid the selection of compounds with a decrease risk of cardiotoxicity in rats. Duplicating the treatment paradigms used in a clinical setting, or extrapolating toxic dose data was not the goal of these preliminary studies. Rather the goal was to determine if single dose, resource-sparing study design could facilitate prioritization of compounds before progressing to more labor intensive and costly pre-clinical studies.
Some compounds will not produce immediate histological evidence of cardiac toxicity, relying on multiple doses or lengthy time periods before histological or biochemical changes are evident. Consequently, the fact that no lesions or traditional biochemical markers were detected in the DIG, DOX, ISO treated rats in this study is not surprising and emphasizes the ability of toxicogenomic methods to predict toxic liabilities early during the safety screening process. Cardiac lesions in the LPS and AG treated rats were clearly identified a week after administration, but this did not preclude the fact that these two compounds displayed significant changes in cardiac tissue gene expression at earlier time points.
We applied relatively stringent criteria for our gene list with the aim of increasing specificity and to focus our selections for subsequent Q-PCR analysis. Even less stringent criteria have the potential of including more genes that are possible biomarkers, without weeding out potential genes of interest. There are currently no acceptance criteria or rules establish for such an approach, however, the simple stringency criteria outline in this paper facilitates selection of a biomarker gene list that can be rapidly applied to early discovery programs and quickly measured using Q-PCR. Intuitively, these criteria should be stringent enough to reduce false-positive (toxicity) results, but inclusive enough to identify a wide range of cardiotoxic compounds.
In summary, this study documented a set of 17 genes that change in expression in rats after treatment with cardiotoxicants that differ in their mechanisms of toxicity. Though these represent only preliminary findings, a more developed list of gene expression biomarkers of cardiac toxicity could contribute to early prioritization of potential new drugs. Future studies could capture xenobiotic induced transcriptome wide changes in gene expression across time, in a database that includes a multitude of compounds with an assortment of appropriate negative controls. After biomarkers genes are substantiated, other methods such as quantitative PCR could substitute for GeneArray analysis, saving money and allowing for the analysis of additional samples, time points and such. A similar set of biomarker genes could be established for other major target organs of toxicity. In cases where expression patterns can be linked to a specific toxic event, expression profiling may provide a faster and less resource-requiring screening strategy for drug discovery projects.