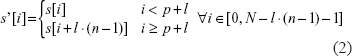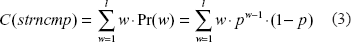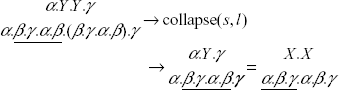In spite of the recognized importance of tandem duplications in genome evolution, commonly adopted sequence comparison algorithms do not take into account complex mutation events involving more than one residue at the time, since they are not compliant with the underlying assumption of statistical independence of adjacent residues. As a consequence, the presence of tandem repeats in sequences under comparison may impair the biological significance of the resulting alignment. Although solutions have been proposed, repeat-aware sequence alignment is still considered to be an open problem and new efficient and effective methods have been advocated. The present paper describes an alternative lossy compression scheme for genomic sequences which iteratively collapses repeats of increasing length. The resulting approximate representations do not contain tandem duplications, while retaining enough information for making their comparison even more significant than the edit distance between the original sequences. This allows us to exploit traditional alignment algorithms directly on the compressed sequences. Results confirm the validity of the proposed approach for the problem of duplication-aware sequence alignment.
Pairwise sequence alignment algorithms are based on metrics derived from edit distance,1 which share the assumption of statistical independence among the single-nucleotide mutation events used to explain the differences between the two sequences under comparison.
Unfortunately, this assumption doesn't hold in case of tandem duplications which involve more than one nucleotide at the time, resulting in the so-called tandem repeats (TRs). The application of traditional alignment algorithms to sequences containing TRs might lead to alignments which are not biologically sound.2 Since the impact of these events is not marginal, as short tandem duplications ranging from 1 to 100 base pairs account for the majority of insertion events in human genome,3 it is mandatory to develop sequence alignment methods capable of taking them into account as underlying biological mechanisms.
Any TR is the effect of subsequent duplications of a repeat unit, called motif. During the evolutionary process, random mutation events can occur at any time possibly affecting one or more repeat units, making them different from the original motif. Mutated repeat units are called variants, while TRs containing more variants are called approximate TRs. The variability of TRs is not limited to the random mutations of the motif, but it is also caused by DNA replication slippage which alters the number of repetitions.5,6 The high instability of TRs is a well known fact which is exploited by using polymorphic tandem repeat loci (also known as variable number tandem repeats, VNTR) as genetic markers.7 In spite of the worth of taking into account multi-nucleotide duplications during the comparison of biological sequences, even the definition of TR is not universally accepted and it is particularly controversial in case of approximate TRs. The lack of a common agreed definition impacts the detection of TRs and it has determined the development of different detection algorithms4,8–11 leading to different results.12 The regular structure of TRs can be exploited to reduce the size of the representation. For instance, Berard et al use macro-characters to represent each motif (and its variants) and apply run-length encoding (RLE) to the sequence of macro-characters.7 In general, compression techniques have the two-fold objective of reducing memory requirements and speeding up comparison.
There are two levels of granularity at which repeat-aware sequence comparison can be performed. The coarse-grained problem consists of aligning two sequences that possibly contain TRs, the fine-grained problem consists of aligning two TRs. The first problem has been tackled by means of modified dynamic programming algorithms13 with time complexity O(N5) and space complexity O(N2) (or with time complexity O(N4) and space complexity O(N3)), where N is the length of the input strings. Solutions to the second problem have been proposed that allow the comparison of TRs according to different evolutionary events, including operations on single characters and more complex operations on substrings of the given sequences (eg, excisions and rearrangements).7,15 TRs to be used as input for this category of algorithms are usually represented as sequences of macro-characters belonging to a super-alphabet which encode possible variants of a given repeat (for instance, in the representation of minisatellite maps obtained by means of PCR-based methods).16 Within this framework, two solutions have been proposed which differ in the type of evolutionary model they handle which, in turn, impacts algorithmic complexity. Berard et al proposed an algorithm for the comparison of minisatellite maps whose time complexity is O (p3 +|Σ| 3), where Σ is the alphabet, s and r are the original maps under comparison, s and r are their RLE compressed versions, |.| denotes either the cardinality of a set or the length of a sequence, = max (||,||), and p = max (||,||). Sammeth and Stoye introduced an algorithm with time and space complexity exponential in the length of input sequences s and r.15 The exponential complexity is paid for accounting for a more complex evolutionary model where several variants could be duplicated in a single event. In summary, while solutions to the coarse-grained problem incur heavy computational burdens and make simplifying assumptions on the mutation type and order; solutions to the finegrained problem are compatible with more accurate evolutionary models but they rely on the availability of predefined TRs.
Taking a different approach, a common practice (called repeat masking)17 consists of pre-processing the sequences under comparison in order to mask the tandem repeats which might impair the biological significance of the alignment. Traditional algorithms based on edit distance can be applied downstream to the repeat-masked sequences, thus overcoming the issue of repeat-aware alignment.
Since none of the proposed approaches clearly outperforms the others in all possible contexts, a methodology for driving the choice of the most appropriate algorithm to be adopted to tackle a specific problem has been recently proposed.18 It makes use of significance metrics which represent the evolutionary likelihood of the results provided by the candidate sequence alignment algorithms. A Monte Carlo approach can be adopted in order to conduct a sensitivity analysis in the parameter space.
This paper presents a new solution to the coarsegrained problem which resembles repeat-masking techniques, in that it addresses exactly the same issues and it entails a lossy compression mechanism which provides approximate representations of the sequences under comparison. The algorithm aims at reducing the original input sequences (that contain repeats) into sequences without exact TRs (hereafter also denoted as repeat-free sequences) by iterative collapsing of repeats of increasing length. After the overall procedure the sequences retain, up to a given degree, enough informative content for the significance of their comparison, while not being affected by the problem of duplication (as they are repeat-free by construction).
The proposed compression algorithm has O(N3) worst case time complexity and O(N2) average case time complexity. Experimental results show that the new method outperforms both pure edit distance and repeat-masking techniques in terms of quality metrics18 when applied to the alignment of sequences highly affected by duplication events.
Repeat Collapsing Algorithm
This section introduces an iterative algorithm which provably turns a sequence containing TRs into a sequence which doesn't contain any. This opens the way to a new (approximate) representation of the original sequences that can be then compared by means of traditional alignment algorithms, thus circumventing the problem of statistical independence in duplication-rich regions. After having settled preliminary definitions, the repeat collapsing algorithm is outlined and its correctness and complexity are proved. In the following we use TR to implicitly denote, with a slight abuse of notation, exact tandem repeats.
Given a string s, a substring s' of with length l and position p is denoted by s' = s[p, p + l - 1]. A TR with n repeat units of size l starting from position p in s is denoted by TR(s, l, p, n). In symbols
The k-th repeat unit of t is tk = t[k · l,k · l + l - 1] = s[p + k · l,p + k · l + l - 1]. Repeat unit t0 is the template of t. TR t is said to be the first TR with template of length l in s, denoted by fTR(s, l), if there are no TRs of size l in s with position lower than p.
Definition: The collapsing of TR t = TR(s, l, p, n) is the transformation of string s' into a shorter string s (hereafter called residual string) obtained by excising all the repeat units of t but the template. In symbols:
Definition: The size-l collapsing of string s is the transformation of string s into a string s' with no TRs of size l, obtained by iteratively collapsing TRs of size l.
An algorithm for performing the size-l collapsing of a string is shown in Figure 1. The algorithm spans the original string (s) with a sliding window of size l, which points out, at each iteration, the substring to be considered as the putative template of a size-l TR. The putative template at position i is then compared with the substring starting at i + l. If they match, a TR is found and the algorithm looks forward to count the number of occurrences of the repeat unit after the template (k) until a mismatch is encountered. The i-th character of s is then copied in the output string s', and the sliding window is shifted from i to i + 1 + k*l. If a TR was found, then k > 0 and the window is moved to the second character of the last occurrence of the repeat unit, thus causing a single occurrence of the template to be copied in s', according to the collapsing rules of Equation 2. If no TR was found in position i, then the window is shifted by 1 position only. The correctness and complexity of the algorithm are assessed by the following theorem.
Algorithm for obtaining a size-l collapsing of a given string s.
Theorem: Algorithm collapse (s, l) of Figure 1 performs a size-l collapsing of string s with worst-case complexity O(N · l) and average-case complexity O(N), where N is the length of the original string and l is the size of the repeats to be found and collapsed.
Proof [Correctness]. The size-l collapsing of a string s could be iteratively performed by collapsing its first TR of size l, fTR(s, l), and by repeating the process on the residual string until it contains no more TRs. If we denote by s(h) the residual string obtained after h iterations, and by p(h) the position of fTR(s(h), l), than it can be easily shown that p(h) > p(h-1) for any h > 0. In fact, the collapsing of a TR in position p cannot cause the emergence of TRs in the previous substring. Hence, the first p(h-1) characters of residual string s(h) do not need to be processed at step h since they have been collapsed at previous steps. According to this observation, the iterative process outlined at the beginning of this proof can be implemented by parsing the string only once. This is exactly what the collapse(s, l) algorithm does.
Proof [Complexity]. The algorithm spans the original string and performs, at each position, a comparison between two substrings of size l. Since substring comparison stops as soon as a difference is encountered, the number of steps involved in each comparison ranges from 1 to l, depending on the length (w) of the coincident prefixes of the substrings under comparison. If the two substrings are identical, the comparison takes O(l). However, this is not the worst case for the collapse algorithm, since whenever a TR is encountered, the loop counter is incremented by l, saving l iterations. As a consequence, the O(l) complexity of substring comparison can be regarded as distributed over the l skipped iterations, bringing to a constant average complexity per iteration. The true worst case occurs when there are no TRs and the first difference between the two substrings under comparison is found, on average, after l/2 characters, leading to an overall complexity of O(N · l).
Average-case complexity of the comparison between strings of size l can be computed by taking into account the probability of finding the first mismatch in position w, that can be expressed as a function of the probability p of finding two matching characters, under the simplifying assumption of base independence:
where Pr(w) denotes the probability of finding the first mismatch in position w. If we extend to infinity the summation of Equation 3 and we exploit its known sum we obtain a constant upper bound for the complexity of strncmp:
Since the average case complexity of the inner loop has a constant upper bound, the overall collapse algorithm executes in linear time O(N).
Definition: A string s is said to be a TR-free string if it contains no TRs of any size.
Algorithm collapse_all of Figure 2 iteratively invokes collapse (s, l) to remove TRs of increasing size l. The following theorem states that the output it returns is a TR-free string.
Algorithm for the obtaining a TR-free string version of a given string s.
Theorem: Algorithm collapse_all (s) of Figure 2 always returns a TR-free string, with worst-case complexity O(N3) and average-case complexity O(N2), where N is the length of s.
Proof [correctness]. Since we know, from Theorem 2, that collapse (s, l) produces a string with no TRs of size l and it is invoked by collapse all (s) for increasing values of l, here we need only to prove that the execution of collapse (s, l) doesn't cause the emergence of TRs of size j < l in the residual string if the input string contains no TRs shorter than l.
Let's assume, by contradiction, that the input string of collapse (s, l) contains no TRs shorter than l, while the output string contains a TR (denoted by tX) of size j < l. Since tX was not present in the input string, its occurrence has to be regarded as the effect of the collapsing of some TR of size l (say, tY). Let's use X and Y to denote the templates of tX and tY, respectively. Without loss of generality, we consider X as composed of three substrings: α, β, and γ. Using the dot to denote string concatenation we can write X = α. β. γ.
According to our assumption, the output string contains a substring of the form X.X = α. β. γ. α. β. γ. that was not in the input string because of the presence of tY. Since the collapsing of tY has the only effect of reducing a substring of the form Y. Y ….. Y to a substring of the form Y, we start from the result of collapsing (i.e. X.X) and we look for a suitable template Y the duplication of which has the effect of masking tX.
Notice that Y has to be contained in X.X in order for the collapsing of tY to affect tX. Hence, we restrict our search of Y among the substrings of X.X. Moreover, we know that Y is longer than X. The only candidate solution is Y = β. γ. α. β, which may cause an original string containing a substring of the form α. Y. Y. γ to become X.X after collapsing:
Notice, however, that the input string contains two adjacent copies of substring β, which form a TR of size |β| < l: a contradiction.
Since the above example is representative of all possible cases, there are no size-l TRs the collapsing of which can cause the emergence of a TR of size j < l starting from a string with no TRs shorter than l.
Hence, if collapse (s, l) is first invoked for l = 1 and then iteratively invoked for increasing lengths, the result is a TR-free string.
Proof [complexity]. Worst-case and average-case complexities come from those of collapse (s, l), which is repeatedly invoked by collapse all (s) for l ranging from 1 to the maximum size of the possible repeat units, which is upper bounded by N/2.
In the worst case, no TRs are found and the inner procedure is invoked N/2 times, so that the overall complexity is given by the following sum:
In the average case, some TRs are found, so that the length of the residual string decreases over time and the number of iterations of the main loop is lower than N/2. However, both the length of the residual strings (which determines the average-case complexity of collapse (s, l) according to Theorem 2) and the number of iterations are still proportional to N, so that the overall complexity is O(N2). The behavior of algorithm collapse_all is exemplified in Figure 3A, which shows a sequence of 36 bases containing several nested repeats, which is reduced to a repeat-free sequence of 11 bases in three collapsing steps for repeat lengths ranging from 1 to 3.
Example of repeat collapsing performed by: (A) algorithm collapse_all of Figure 2, (B) algorithm collapse of Figure 1 itera-tively invoked for repeat lengths ranging from 3 to 1, and (C) algorithm collapse iteratively invoked for lengths ranging from 5 to 1.
Figure 3B shows, for comparison, the result that would be achieved by invoking the collapse algorithm in reverse order, ie, for repeat length decreasing from 3 to 1. It is apparent that the resulting string is not repeat-free. In fact, it is of 17 bases and it contains three replicas of the acg motif.
Even starting from a longer repeat length the reverse collapsing procedure does not guarantee to remove all tandem repeats. This is shown in Figure 3C, which starts from repeat length 5 (which is the length of the longer motif found in the original sequence) to achieve a collapsed sequence of 14 bases with two replicas of the acg motif.
Observation
We point out here that the identification of exact TRs could be solved by means of suffix trees data structures very efficiently with O(N2logN), being N the length of the string and z the number of occurrences of the TRs.19 In principle, our approach could also be extended to take advantage of these results. Notice however that we need to collapse tandem repeats iter-atively, starting from the shortest ones, and the search for longer TRs has to be repeated at each step (in fact, collapsing of short TRs can determine the emergence of longer ones). Hence, the benefits coming from the usage of suffix trees could be exploited only within the collapse procedure, which is invoked O(N) times within the inner loop of collapse all, leading to an overall time complexity of O(N2logN), which is better than the worst-case complexity of the proposed algorithm, but worse than its average-case complexity.
It is also worth noticing that the computational efficiency of collapsing is not the main concern, since in a database search setting entries could be compressed off-line once and for all, thus reducing the impact of collapsing algorithm on runtime performance. Notably, an interesting by-product of our approach is the possibility of reducing the complexity of a query search in the database by virtue of compression.
Results and Discussion
The compression algorithm described so far has been conceived to be applied as a pre-processing step in order to increase the performance of traditional alignment algorithms based on edit distance when applied to sequences rich of TRs. The combined application of repeat collapsing and edit distance is hereafter denoted by RC.
The benefits of the RC approach have been evaluated by following a specific methodology recently introduced to assess the quality of repeat-aware alignment algorithms.18 In particular, three metrics have been used to quantify the biological-significance of the results provided by the alignment algorithm:
significance ratio (R), which is the ratio between the number of aligned bases/residues coming from the same base/residue of a common ancestor, and the length of the shortest of the two sequences under alignment;
selectivity (S), which is defined as the probability for a database entry se to be ranked first by the alignment algorithm used to search among the M entries of a database with a query string s provided that se is the only entry in the database homologous to sq;
ranking error (E), which is the normalized position of se in the ranking produced by the alignment algorithm when sq is used as a query: E = (rank(se) - 1)/(M- 1).
The three metrics are to be evaluated on a set of synthetic benchmarks randomly generated by means of a Monte Carlo approach which simulates the evolution process according to the following statistical parameters: the probability of single-nucleotide insertion (pins), deletion (pdel), and mutation (pm), the probability of duplication (pd), the probability of extension of an existing TR (pe), the maximum size of a repeat unit (L), the number of evolution epochs (T), the number of known ancestors generated at each run (M), and their average length (N).
In order to test the proposed RC approach against a significant set of Monte Carlo experiments, we applied all the settings that were originally used to test the sensitivity of the quality metrics.18 In particular, we run 200 Monte Carlo experiments in the neighborhood of a representative point of the parameter space (hereafter called baseline) summarized in Table 1. It is worth noticing that the computation of the quality metrics entailed 8,000,000 pairwise alignments of sequences of about 200 bases each.
Ranges of the parameters used for Monte carlo simulations.
Parameters
M
T
pins
pdel
pd
pe
pm
L
N
Min
160
40
0.00008
0.00008
0.0008
0.004
0.0008
12
80
Max
240
60
0.000012
0.000012
0.0012
0.006
0.0012
18
120
Avg
200
50
0.000010
0.000010
0.0010
0.005
0.0010
15
100
Abbreviations: M, number of ancestral DNA sequences; T, number of epochs considered as evolution time; pins/pdel/pd/pe/pm, insertion/deletion/duplication/extension/mutation probabilities; L, maximum size of a repeat unit; N, length of the ancestral DNA sequences.
The adoption of this experimental setup provides the additional advantage of making our results directly comparable with those of the alternative approaches used as a case study in the paper were the quality metrics were originally introduced,18 namely, bare edit distance (ED) and different flavors of repeat masking (RM) performed by mreps.9
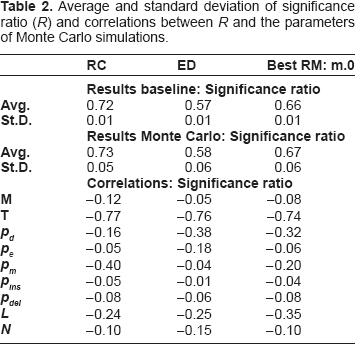
Comparative results are provided in Tables 2–4. Column labeled “RC” refers to the proposed approach, column “ED” refers to bare edit distance, while column “Best RM” refers to the repeat masking technique which provided the best results according to previous work.18 In order to enable a thorough evaluation of the sensitivity of the results from the parameters adopted to run Monte Carlo simulations, Pearson correlations were also computed and reported in the results tables.
Average and standard deviation of significance ratio (R) and correlations between R and the parameters of Monte carlo simulations.
RC
ED
Best RM: m.0
Results baseline: Significance ratio
Avg.
0.72
0.57
0.66
St.D.
0.01
0.01
0.01
Results Monte Carlo: Significance
Avg.
0.73
0.58
0.67
St.D.
0.05
0.06
0.06
Correlations: Significance ratio
M
-0.12
-0.05
-0.08
T
-0.77
-0.76
-0.74
pd
-0.16
-0.38
-0.32
pe
-0.05
-0.18
-0.06
pm
-0.40
-0.04
-0.20
pins
-0.05
-0.01
-0.04
pdel
-0.08
-0.06
-0.08
L
-0.24
-0.25
-0.35
N
-0.10
-0.15
-0.10
Average and standard deviation of selectivity (S) and correlations between S and the parameters of Monte carlo simulations.
RC
ED
Best RM: m.n2
Results baseline: selectivity
Avg.
0.89
0.75
0.87
St.D.
0.03
0.02
0.03
Results Monte carlo: selectivity
Avg.
0.89
0.73
0.87
St.D.
0.05
0.09
0.06
Correlations: selectivity
M
-0.16
-0.12
-0.12
T
-0.61
-0.68
-0.71
pd
-0.20
-0.30
-0.32
pe
-0.02
-0.18
-0.13
pm
-0.31
-0.02
-0.08
pins
-0.01
0.02
0.04
pdel
-0.06
-0.03
-0.09
L
-0.46
-0.44
-0.37
N
0.33
0.22
0.22
Average and standard deviation of ranking error (E) and correlations between E and the parameters of Monte carlo simulations.
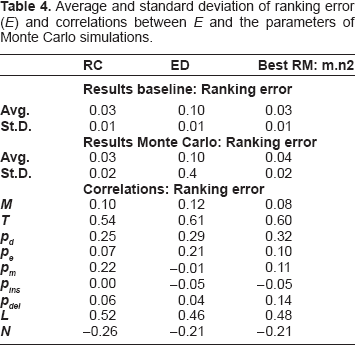
RC
ED
Best RM: m.n2
Results baseline: Ranking error
Avg.
0.03
0.10
0.03
St.D.
0.01
0.01
0.01
Results Monte carlo: Ranking error
Avg.
0.03
0.10
0.04
St.D.
0.02
0.4
0.02
Correlations: Ranking error
M
0.10
0.12
0.08
T
0.54
0.61
0.60
pd
0.25
0.29
0.32
pe
0.07
0.21
0.10
pm
0.22
-0.01
0.11
pins
0.00
-0.05
-0.05
pdel
0.06
0.04
0.14
L
0.52
0.46
0.48
N
-0.26
-0.21
-0.21
Table 2 shows that RC significantly outperforms both ED and Best RM in terms of significance ratio (0.73 on average for RC versus 0.58 and 0.67 of ED and RM respectively). Best RM in Table 2 refers to the masking obtained by filtering out all exact TRs including the small ones (mreps parameter settings res = 0 and allowsmall = true, denoted by m.0).18,9 The higher robustness of RC against TRs is also demonstrated by its lower sensitivity to the probabilities of duplication (pd) and extension (pe). On the other hand, the quality of the results provided by RC is highly affected by mutation probability (pm) since it might reduce the effectiveness of collapsing by changing exact TRs into approximate ones which are not targeted by the proposed approach. Notice however that the high sensitivity to mutation probability is common to all repeat-aware techniques, including RM which shows a correlation to pm higher than ED.
Table 3 shows that the selectivity (S) of RC is remarkably higher than that of ED and slightly higher than that of Best RM (0.89 on average for RC versus 0.75 and 0.87 for ED and RM, respectively). In this case Best RM refers to the masking obtained by filtering out all approximate TRs up to resolution 2 but the small ones (this corresponds to mreps settings res = 2 and allowsmall = false, denoted by m.n2).18,9 From the correlation analysis we can observe that the spread between RC, ED, and Best RM in terms of sensitivity grows as duplication and extension probabilities increase (pd, pe), while it reduces for larger values of mutation probability (pm).
Finally, Table 4 shows that RC significantly outperforms ED in terms of ranking error (E), while it performs slightly better than Best RM, which refers to the same masking strategy which was used as a term of comparison in Table 3. As already mentioned for R and S, RC has the lowest sensitivity to the probabilities of duplication (pd) and extension (pe), while it is more sensitive to mutation probability (pm). In this case it is also worth noticing a lower sensitivity to the number of evolutionary epochs (T).
The results presented in this section clearly provide the evidence of the improved quality of RC-based alignments in terms of R, S, and E. Interestingly, RC is a lossy compression technique which can also provide benefits in terms of computational and memory complexity. Most important, the improved quality of the alignments obtained from TR-free sequences demonstrates that the compression technique has the capability of retaining the substrings which are more significant for the alignment.
Statistical significance
The statistical significance of the comparative results provided in Tables 2–4, was assessed by performing the Wilcoxon signed rank test14 on the outcomes of the 200 Monte Carlo experiments. The test returns a so-called P-value which represents the probability for the pairwise difference between two approaches (as returned by the Monte Carlo experiments) to be only explained by random sampling rather than by the actual difference in the distributions of their parent populations. Hence, the lower the P-value the higher the statistical significance of the comparative results.
The results of the Wilcoxon test are reported in Table 5. All the P-values were lower than 10−19, demonstrating the statistical significance of the experiments.
Results of Wilcoxon signed rank test.
Metrics
ED vs. RC
RM vs. RC
R
1.432E-34
1.432E-34
S
1.434E-34
2.253E-11
E
1.436E-34
3.297E-19
Further evidence of the different performance of the three methods is provided by the scatter plots of Figures 46, which report, for each metric, the value achieved by ED and RM, plotted against those achieved by RC. The solid line in each graph represents the bisector of the Cartesian plane, plotted as a reference, while dashed lines represent the linear regressions of the corresponding sets.
Scatterplot of ED vs. RC and RM vs. RC in terms of significance ratio.
Figure 4 clearly shows the superior quality of RC in terms of significance ratio, since all the points are well below the bisector. Figure 5 confirms that the selectivity of RC is much higher than that of ED (the points of which are all below the bisector) and slightly, but consistently higher than that of RM. Finally, Figure 6 shows that the ranking error of RC is much lower than that of ED (the points of which are all above the bisector) and slightly lower than that of RM.
Scatterplot of ED vs. RC and RM vs. RC in terms of selectivity.
Scatterplot of ED vs. RC and RM vs. RC in terms of ranking error.
Biological dataset experiments
In order to evaluate the capability of the proposed approach of leading to significant alignments between biological sequences, we also tested it on real data. To this purpose we used a dataset commonly adopted for benchmarking,20,21 composed of the mitochondrial DNA sequences of 15 species with lengths ranging from 16295 bp (Mus musculus) to 16797 bp (Halichoerus grypus). Both the RC and ED alignment methodologies (as defined in Section 3) were applied to compute the pairwise distances among all the 15 sequences. The resulting distance matrices were then used to derive phylogenetic trees by means of Neighbor Joining.22 In the following we use RC-tree and ED-tree to denote the results obtained with and without repeat collapsing.
In order to evaluate the quality of the phyloge-netic trees we computed their Robinson-Foulds distance (RFd)23 from the gold standard adopted by Ferragina et al21 for the same data set. The RFd is a metric commonly used in computational phy-logenetics for the topological comparison of trees. The value it takes is defined between 0 and 4n - 10, where n is the number of taxa of the trees under comparison and 0 corresponds to isomorphic trees. For our benchmark RFd was defined in the [0,50] interval and was computed by means of the tree-dist-sym-dif.pl Perl script provided in the Kolmogorov Library.21 The distance from the gold standard resulted to be 3 for RC-tree and 9 for ED-tree, confirming the capability of the collapsing strategy to reduce the noise (caused by the improper assumption of statistical independence of adjacent bases) which might impair the results of sequence alignment in presence of TRs.
It is worth noticing that RFd = 3, which is the partition distance from the gold standard achieved by the RC-tree, is in line with the best results achieved by compression-based techniques.21 In particular, only 2 out of the 75 techniques tested by Ferragina et al obtained RFd = 3, while all others obtained partition distances between 5 and 23 (see Table 6 of Ferragina et al).21
Needless to say, this is a simple case study which doesn't provide any general validation. Rather, it complements the results already obtained in terms of significance metrics computed on synthetic benchmarks, as discussed in Section 3.
Conclusions
This paper has presented a new approach to the problem of duplication-aware sequence alignment. The proposed method hinges upon a preprocessing that takes original sequences and computes a repeat-free version of them. Directly working on such approximate representation provides the attractive advantage of speeding up comparison while increasing edit-distance significance because of the enhanced properties of statistical independence between adjacent residues (which is usually impaired by duplication events).
An efficient algorithm for repeat collapsing has been presented and its properties have been formally proved. The capability of the proposed approach to enhance the quality of pairwise alignments has been evaluated in terms of the significance metrics recently introduced to assess the quality of duplication-aware alignment algorithms. Comparative results obtained from Monte Carlo simulations have confirmed the effectiveness of the proposed approach, which has also proved its effectiveness in reconstructing a phy-logenetic tree starting from a biological dataset.
Author Contributions
Conceived and designed the experiments: VF, AB. Analysed the data: VF, AB. Wrote the first draft of the manuscript: VF, AB. Contributed to the writing of the manuscript: VF, AB. Agree with manuscript results and conclusions: VF, AB. Jointly developed the structure and arguments for the paper: VF, AB. Made critical revisions and approved final version: VF, AB. All authors reviewed and approved of the final manuscript.
Footnotes
As a requirement of publication author(s) have provided to the publisher signed confirmation of compliance with legal and ethical obligations including but not limited to the following: authorship and contributorship, conflicts of interest, privacy and confidentiality and (where applicable) protection of human and animal research subjects. The authors have read and confirmed their agreement with the ICMJE authorship and conflict of interest criteria. The authors have also confirmed that this article is unique and not under consideration or published in any other publication, and that they have permission from rights holders to reproduce any copyrighted material. Any disclosures are made in this section. The external blind peer reviewers report no conflicts of interest.
References
1.
GusfieldD.Algorithms on Strings, Trees and Sequences: Computer Science and Computational Biology.Cambridge University Press, UK, 1997.
2.
MorgensternB., ProhaskaS., PohlerD., StadlerP.Multiple sequence alignment with user-defined anchor points. Algorithms for Molecular Biology.2006; 1(1): 6.
3.
MesserP.W., ArndtP.F.The majority of recent short DNA insertions in the human genome are tandem duplications. Molecular Biology and Evolution.2007; 24: 1190–7.
4.
DelgrangeO., RivalsE.STAR: an algorithm to search for tandem approximate repeats. Bioinformatics.2004; 20(16): 2812–20.
5.
DieringerD., SchlottererC.Two distinct modes of microsatellite mutation processes: Evidence from the complete genomic sequences of nine species. Genome Res.2003; 13: 2242–51.
BerardS., NicolasF., BuardJ., GascuelO., RivalsE.A fast and specific alignment method for minisatellite maps. Evolutionary Bioinformatics.2006; 2(1): 327–44.
8.
BensonG.Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res.1999; 27(2): 573–80.
9.
KolpakovR., BanaG., KucherovG.Mreps: Efficient and flexible detection of tandem repeats in DNA. Nucleic Acids Res.2003; 31(13): 3672–8.
10.
LandauG.M., SchmidtJ.P., SokolD.An algorithm for approximate tandem repeats. J Comput Biol.2001; 8: 1–18.
11.
WexlerY., YakhiniZ., KashiY., GeigerD.Finding approximate tandem repeats in genomic sequences. J of Comput Biol.2005; 12(7): 928–42.
12.
LeclercqS., RivalsE., JarneP.Detecting microsatellites within genomes: significant variation among algorithms. BMC Bioinformatics.2007; 8(1): 125.
13.
BensonG.Sequence alignment with tandem duplication. J Comp Biol.1997; 4: 351–67.
14.
RosnerB., GlynnR.J., LeeM.L.T.The wilcoxon signed rank test for paired comparisons of clustered data. Biometrics.2006; 62(1): 195–92.
15.
SammethM., StoyeJ.Comparing tandem repeats with duplications and excisions of variable degree. IEEE/ACM Transactions on Computational Biology and Bioinformatics.2006; 3(4): 395–407.
16.
JeffreysA.J., MacLeodA., TamakiK., NeilD.L., Monck-tonD.G.Minisatellite repeat coding as a digital approach to DNA typing. Nature.1991; 354: 204–9.
17.
ClaverieJ.M.Computational methods for the identification of genes in vertebrate genomic sequences. Human Molecular Genetics.1997; 6(10): 1735–44.
18.
FreschiV., BoglioloA.A monte carlo method for assessing the quality of duplication-aware alignment algorithms. Evolutionary Bioinformatics.2011; (7): 31–40.
19.
StoyeJ., GusfieldG.Simple and flexible detection of contiguous repeats using a suffix tree. Theoretical Computer Science.2002; 270: 843–56.
20.
ApostolicoA., CominM., ParidaL.Mining, compressing and classifying with extensible motifs. Algorithms for Molecular Biology.2006; 1(1): 4.
21.
FerraginaP., GiancarloR., GrecoV., ManziniG., Va-lienteG.Compression-based classification of biological sequences and structures via the Universal Similarity Metric: experimental assessment. BMC Bioinformatics.2007; 8(1): 252.
22.
DurbinR., EddyS., KroghA., MitchinsonG.Biological Sequence Analysis.Cambridge University Press, UK, 1998.
23.
RobinsonD.F., FouldsL.R.Comparison of weighted labelled trees. In: Proc. 6th Australian Conf. Combinatorial Mathematics, Lecture Notes Mathematics.1979: 119–26.