
Abstract
Domain Fusion Analysis takes advantage of the fact that certain proteins in a given proteome A, are found to have statistically significant similarity with two separate proteins in another proteome B. In other words, the result of a fusion event between two separate proteins in proteome B is a specific full-length protein in proteome A. In such a case, it can be safely concluded that the protein pair has a common biological function or even interacts physically. In this paper, we present the Fusion Events Database (FED), a database for the maintenance and retrieval of fusion data both in prokaryotic and eukaryotic organisms and the Software for the Analysis of Fusion Events (SAFE), a computational platform implemented for the automated detection, filtering and visualization of fusion events (both available at: Error! Hyperlink reference not valid.). Finally, we analyze the proteomes of three microorganisms using these tools in order to demonstrate their functionality.
Introduction
Protein-protein interactions are of great importance in almost every level of cell function: in DNA replication and transcription, regulation of gene expression, metabolic pathways, signaling pathways, structure of sub-cellular organelles, cell cycle control, to name a few. 1 Understanding the nature of these interactions helps us make inferences for several complex biological processes.
Protein-protein interaction data have been traditionally collected through biochemical and genetic approaches, including the well known “yeast two-hybrid assay”.
2
Marcotte
The complexity of protein interactions and their significance to biological research has intensified the necessity to develop databases storing related information. Representative examples constitute databases such as STRING
6
and DIP
7
dedicated to that specific purpose. For a further detailed investigation of protein relationships, PROLINKS database
8
provides information based on the phylogenetic profiles method, the gene neighbors method, the gene cluster method and the Rosetta Stone method. The latter one led to the development of FusionDB,
9
a specialized database containing fusion events detected in genomes of the archaea and bacteria. Given the wide interest in that particular research field, FED extends and makes available information over fusion events based on bibliographical, computational or
Numerous sequence alignment computational tools are available, which offer visualization of sequence alignments as well. More specifically, there is DnaSP 10 which conducts alignments of nucleotide sequences and visualizes the results generated. In a more protein-centric fashion, Artemis Comparison Tool 11 and Geneious Pro 12 make available both the alignment results and their graphical representation. However, due to FED's particular research aspects, the need for a specialized Fusion Events Extraction and Visualization Application became mandatory. Hence, this led to the implementation of SAFE which aims to handle the automated detection and filtering of fusion events and provide FED with complementary computational research data. Although a number of studies with results from gene fusion analysis have been published, no specific tool for gene fusion analysis is currently available publicly. For this reason, we decided to develop SAFE which has a simple user interface and gives consistently reliable results.
Design and Implementation
Features
Our approach is to infer physical interactions or functional links between proteins, from a computational perspective, by identifying fusion events from sets of amino-acid sequences. FED comprises results either derived from the bibliography, or extracted with the use of our computational tool, which share the same theoretical basis: the fact that certain proteins in a given species consist of fused domains that correspond to a single structural domain or a full-length protein in another species.
Concerning the bibliographically retrieved events, the initial step in the whole procedure was to collect information about fusion events from the scientific literature. The next step was the amino acid sequence retrieval from the source databases. That data mining process determined a further categorization of the gene fusion events involved in the database. The first category consists of results where both component and composite protein information is noted down in the relative scientific articles. The bibliographic analysis was followed by computational verification of the results. The second category comprises results where only the information about the composite protein was fully provided. That is, information about the component proteins participating in a specific fusion event was limited to the name of the respective coding gene. Alternatively, only each protein's functionality was given. In order to detect those specific fusion events, the use of alignment tools (such as BLAST) was mandatory. The third category includes fusion proteins detected computationally, as the respective scientific articles supplied details solely describing the component proteins participating in a fusion event. Finally, the aforementioned tasks conducted throughout the research procedure, led to novel in silico-detected fusion events that were included as well in the database, forming the fourth category. Gene fusion results generated by SAFE are listed in that category, too. Of the 385 events total, included in FED, 101 belong to the first category, 43 belong to the second category, and 43 belong to the third category. Finally, 198 events belong to the fourth category, which represent 14 protein families.
The crucial feature FED possesses is that each fusion event was subjected to individual examination and evaluation before its inclusion in the database. The evaluation of each domain fusion prediction was executed with respect to E-value and identity scores reported by BLAST analysis of the proteins involved in a fusion event. More specifically, computationally analyzed fusion events with an identity score under a threshold of 27% were excluded from the result set; below that level, homology cannot be safely concluded. 13 For novel gene fusion results detected by SAFE, backwards BLAST comparison was conducted as well, in order to guarantee the reliability of fusion events. 9 Aiming to enhance the validity of the results, the overlap between the BLAST hits of both query proteins when aligned to the reference protein must not exceed a number of 35 amino acids.
The application comprising the purely computational part of our research, SAFE, uses the following method to conduct gene fusion prediction. Initially, FASTA files comprising the proteomes of the organisms under analysis are downloaded and processed producing sequence sets of non-identical protein sequences. In the following step, the files are subjected to successive pair-wise proteome comparisons, in an all-against-all protein alignment fashion. Once the BLAST alignments are produced, they are refined according to user-specified parameters (see Features, below). The alignments are then examined and collected, in order to form the primary set of fusion events. The exclusion of protein fusion predictions where multiply-occurring proteins participate follows (see SAFE filtering options below). Then, the remaining fusion events set undergoes a scoring scheme based on a user-selected Expectation Value threshold. At this point, the final fusion results are at the user's disposal. In order to accelerate the researcher's task, two additional fusion files are generated. They present the results where the participating proteins appear exactly once and twice respectively.
FED Query Options
FED contains 385 fusion events detected in 129 different organisms. Two main search axes are provided by the database. The first one enables the user to search by organism name, whilst the second one provides fusion results through search by protein name. More specifically, in the respective search field, users may insert the name of the organism they are interested in. Consequently, they are able to access all the fusion events available where this particular organism participates as the reference or the target proteome. In case the name of the organism is not fully specified by the user, the interface allows successive navigation to a list of organisms whose name contains the characters inserted in the search field. An additional feature provided is the search of a particular organism in alphabetical order, where a list of organisms starting with a specific alphabetical character is at the user's disposal. From the generated list, the user can select a certain organism name, in which he/she will search for proteins participating in putative fusion events. The second category of queries is protein centric. In particular, users can search by protein name and access a collection of synonymous proteins, from where they are able to select the one of interest. In order to enhance the query power, a combined search can be carried out; the web-based platform supports a combination of protein name and taxonomy information as input. Users are allowed to select whether they search for available fusion events where the participating protein exists in archaea, bacteria or eukaryotes.
The main advantage of FED is that the simplicity of its interface minimizes drastically the effort needed to access fusion events data. Moreover, navigation through the web-based platform is straightforward and self-explanatory. Thus, all the above described queries enable users to fully exploit at every step the information provided in the database (Fig. 1).
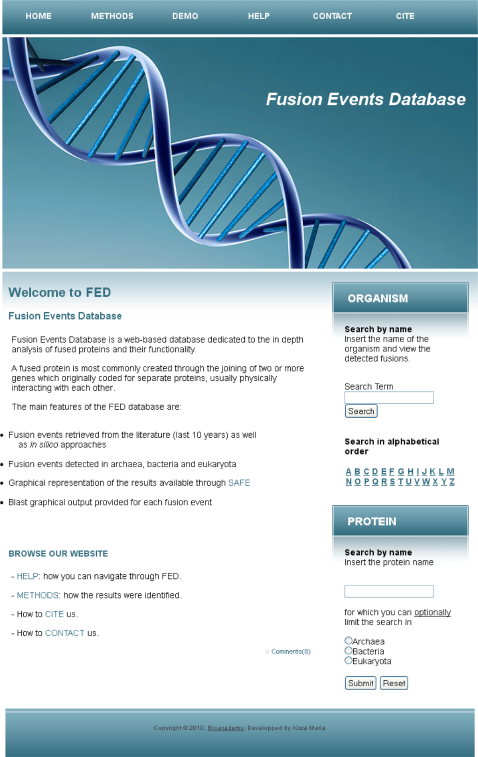
Screenshot of the FED database homepage, showing the available search options.
SAFE Filtering Options
SAFE is designed and implemented with a single main perspective: the adaptability to the user's demands. In other words, users' preferences are incorporated in the automated detection of fusion events. This is accomplished by introducing a set of user-specified parameters, described below, which are incorporated into the workflow of the program as shown in Figure 2.
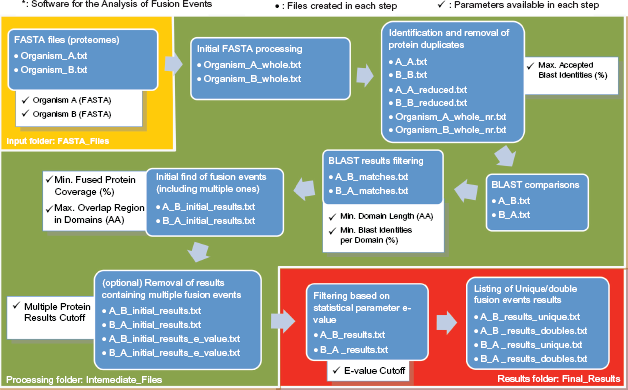
Workflow of the SAFE software. Starting with the input FASTA files of the proteomes of the two organisms that the user wants to analyse, user-specified parameters are used in different steps to filter the data, as described in the text. The output files of the program can be downloaded as text files or visualised on the SAFE interface, as shown in Figure 3.
It is an extremely common phenomenon that the proteome of a particular organism comprises duplicated proteins. To guarantee the integrity of the results the application provides an extra option to exclude redundant proteins from each proteome. Hence, the first parameter sets the threshold required to eliminate the aforementioned proteins.
The next parameter available is called
Another parameter provided allows the user to define the minimum percentage of identities between the protein domains participating in a putative fusion event.
The more extensive the coverage of the fusion protein by the two corresponding component protein domains, the more substantiated a fusion event is. However, the application gives the user the option to set a value
In tandem with the aforementioned requirement, it is of primary importance that both protein domains participating in a fusion event occupy discrete spaces in the respective fusion protein's amino acid sequence. The optimal case would be the absence of any overlap between them. Nevertheless, there are numerous cases of fusion events where some overlap does occur. In order to include those putative fusion events in the procedure, but also to give the user the opportunity to control the number of amino-acids in the overlapping region, the parameters' panel features an additional constraint, the
There are certain cases where a specific protein participates in multiple alignment results. This usually signifies “promiscuous” or paralogous domains, which occur at a high frequency in many different protein sequences that do not share similar functions.15,16 Those proteins are excluded from the results, along with the respective alignments, when the number of their occurrences exceeds the value of
Of course, the E-value holds the leading role among the parameters set both in fusion events extraction and filtering. Hence, it could not be missing from the Project Options panel. The user may set in the respective field the desirable E-value and consequently determine the possibility that a fusion event is valid. The default is set to e-3.
Despite the fact that the parameters are numerous to guarantee optimal results, SAFE presents an extremely user-friendly interface. All the user has to do is drag and drop the input data and he/she is one click away from setting the parameters and starting the fusion events detection process (Fig. 3).

The SAFE interface. On the bottom right is the graphical representation of the selected results.
Results
Results Generated by SAFE
After the Fusion Event Extraction process has finished, SAFE provides the user with the respective results. Aiming to maximize the efficiency of the whole process, we developed SAFE in a manner that enables the researcher to view the results either as a whole, or individually for each fusion event. To achieve this, the platform generates a text file for each organism-against-organism analysis, which contains all the detected fusion events that satisfy the user-entered criteria. In detail, for each fusion result, SAFE offers the user information over the Query and Subject organisms and proteins, their respective alignment regions and also a detailed alignment at the amino acid level. Furthermore, BLAST generated score values are also included, like the Identities, E-value and Positives scores and the number of Gaps (Fig. 3).
When a results file is opened in SAFE, a table containing the respective data is automatically populated. Hence, the user is offered a source of summarized fusion events data, where all the extracted results are presented, each one occupying a table row. In this way, the user can be informed about the respective scores of each fusion event, conduct a comparison between them, if that is needed, and select the desirable ones (Fig. 3).
SAFE'S main attribute is to produce graphical representations of putative fusion events. Each visualization result includes all the substantial information for the respective fusion event. More specifically, this information constitutes the E-value and Identities scores, and the names of both organisms and proteins participating in a fusion event. Each figure aims to offer a simple and straightforward graphical overview that will assist the researcher in a prompt and efficient evaluation for his/her final selection process. To view the graphical representation of a putative fusion event, all the user has to do is click on the respective row (of the desirable fusion event) in the results table mentioned above, and the visualization image will be automatically generated (Fig. 3).
Case Study Conducted via SAFE
The proteomes of two prokaryotic microorganisms, the Archaeon,
We performed our analyses by setting SAFE user-specified parameters' thresholds as follows: Max. Accepted Blast Identities set to 85%, Minimum Domain Length to 80 amino acids, Min. Blast Identities per Domain to 27%, Min. Fused Protein Coverage to 70%, Max Overlap Region in Domains to 0, Multiple Protein Results Cutoff to 2 and E-value to 0.001. With these parameters specified, the platform's fusion event detection algorithm was executed and the following analysis schemes were performed;
Fusion events generated via SAFE.

The novelty of the results can be supported by presenting four specific occasions that occurred, when aligning the proteomes mentioned. In all four cases, two protein domains of each one of the prokaryotic proteomes analyzed (each domain belonging to a separate protein), are found fused in a single, whole-length protein in the eukaryote or the other prokaryote. We suggest that two cytidyltransferase domains of

Examples of fusion events detected by SAFE. (A) A protein from the species
We have additionally identified protein-protein interactions in the
Importantly, for all of the fusion events described, component protein candidates have related biological functions, ie, participate in a common structural complex, metabolic pathway, or biological process. 3
Representation of Results in FED
The fusion events identified by our analysis using SAFE can be searched through the Fusion Events Database. Before accessing the final web page of fusion results, the user navigates through the intermediate search pages that contain information about organisms or proteins participating in fusion events. As far as the organisms are concerned, users can have direct access to taxonomy information via a cross-reference link to UniProt. 20 Additionally, every protein record appears with a link to the corresponding web page in GenBank, 21 where information over its amino acid sequence or its further structural features is available. In the final web page of fusion results all the necessary information about a fusion event is at the user's disposal. That information consists of the proteins that participate in a specific fusion event, with the respective links to GenBank, as described above for both reference and target organisms. FED also provides users with the alignment results corresponding to each fusion event. Those results are generated with the use of BLAST, either via the respective web-interface or via SAFE, when each of the component proteins is aligned to the target one. Apart from the alignment itself, information over the identities and E-value scores is included, providing the necessary biological verification. Furthermore, a novel characteristic is the information provided about any relevant bibliographic resource. The result page comprises the title of the corresponding article, the names of the authors and the scientific journal the article was published in; in case the user wishes to gather more detailed information, the results page features the respective accession number of the article in the PubMed database, as a hyperlink. As has been described in the Methods, the fusion events included in FED are categorized according to the data mining procedure that preceded their further investigation. Hence, concise information about the category of each fusion event included in the database is provided as well.
Availability and Future Directions
Both the Fusion Events Database (FED) and the Software for the Analysis of Fusion Events (SAFE) can be found at the following address: http://www.bioacademy.gr/bioinformatics/projects/ProteinFusion/index.htm. The software mentioned runs on Matlab but will also be available soon in a Java version. One future goal for this project is to make SAFE run faster, by dividing the jobs submitted to run, to a computer cluster or to different servers that offer higher computing capabilities.
Discussion
FED is a stand-alone platform implemented for the analysis of fusion proteins and their functionality, which contains more proteins/organisms and more detailed annotations, compared to previous relevant work. It comprises 385 fusion events, providing detailed information about the proteins each one consists of. Those proteins are carefully aggregated and thoroughly investigated via tools of computational biology, in order to provide substantial verification of each fusion event. Moreover, the fusion events included in FED are reported by journal articles released in the last ten years. A thorough bibliographic research preceded data retrieval and further data computational investigation. Consequently, information over bibliographic resources is also provided, in tandem with the corresponding alignment results generated by NCBI blast. Besides being a curated database, FED also extends previous fusion databases (e.g. FusionDB) 9 by including fusion proteins detected not only in archaea or bacteria but in eukaryotic organisms as well (Supplementary Table 1). Moreover, particular cases, where more than two proteins (or their respective structural domains) participate as components in a fusion event corresponding to a single fusion composite protein, are available. Crucially, the platform also features graphical representation of results generated exclusively by SAFE.
SAFE is a standalone innovative application implemented for the automated detection, filtering and visualization of fusion events. It conducts pairwise alignments among protein sets derived from complete genomes, using user-specified parameters. Through SAFE, the process of in-depth analysis of fusion proteins is simplified and highly accelerated, providing optimum results.
22
The performance of the software was tested against a previous benchmark study of gene fusions,
4
which showed that the results generated by SAFE agree with other methods but the software is also highly selective.
22
SAFE detected almost 90% of the events reported by Enright
As with any automatic analysis, results generated by SAFE can be filtered and analysed further to extract meaningful biological conclusions. For example, once a fusion event is detected in one organism, BLAST can be used to search for the component proteins in other organisms, to check if the protein pair exists as two separate proteins, or is encoded by one fused ORF. This analysis can be extended to cover multiple lineages, to generate a phylogenetic profile of the fusion event, and pinpoint the timepoint during evolution when the fusion or fission event occurred.22,23 Importantly, one should check if the component proteins identified are located adjacent to each other in the genome, as this may point to mis-annotations, leading to artifacts, i.e. not true fusion events. In such cases further checks, e.g. for synteny with closely related genomes can be used to check the annotation, and confirm the fusion event.
Authors' Contributions
DT designed the SAFE software, MK developed the FED database, VD and PT were involved in data analysis and drafting the manuscript and figures. VLK helped with data interpretation and drafting the manuscript. ADK, AT, and SK contributed to the conception and design of the work, and critically revised the manuscript. All authors read and approved the final manuscript.
Supplementary data
Comparison of the search features and the type of data stored in the FED database and in FusionDB. 9

Gene ontologies for the novel fusion events included in the FED databases, i.e. fusion events which were not previously reported in the literature.

Only one accession number is given per protein family.
Footnotes
Acknowledgements
The authors wish to thank Dimitris Dimitriadis for his contribution to the troubleshooting of SAFE, George Kritikos and George Velissaris for helpful discussions on the development of the SAFE, Manolis Balsomatzis for initial work concerning the visualization of the data, Christos Makris for valuable help concerning the database development, and finally Karin Söderman for updating the FED database and also for creating the hosting webpage for the tools presented here. This work was partly supported by the EDGE (National Network for Genomic Research) EU and Greek State co-funded Project (09SYN-13-901 EPAN II Cooperation grant). Amalia D. Karagouni, Vasilis Danos and Sophia Kossida acknowledge the “Heracleitus II” research fellowship program entitled: In silico analysis for microorganisms of medical importance, detection and evaluation of protein interactions and fusion events. Sophia Kossida is a member of the FP7, COST program, “Next Generation Sequencing Data Analysis Network”. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author(s) have provided signed confirmations to the publisher of their compliance with all applicable legal and ethical obligations in respect to declaration of conflicts of interest, funding, authorship and contributorship, and compliance with ethical requirements in respect to treatment of human and animal test subjects. If this article contains identifiable human subject(s) author(s) were required to supply signed patient consent prior to publication. Author(s) have confirmed that the published article is unique and not under consideration nor published by any other publication and that they have consent to reproduce any copyrighted material. The peer reviewers declared no conflicts of interest.