
Abstract
Protein-protein interactions (PPIs) play a crucial role in the life cycles of living cells. Thus, it is important to understand the underlying mechanisms of PPIs. Although many high-throughput technologies have generated large amounts of PPI data in different organisms, the experiments for detecting PPIs are still costly and time-consuming. Therefore, novel computational methods are urgently needed for predicting PPIs. For this reason, developing a new computational method for predicting PPIs is drawing more and more attention. In this study, we proposed a novel computational method based on texture feature of protein sequence for predicting PPIs. Especially, the Gabor feature is used to extract texture feature and protein evolutionary information from Position-Specific Scoring Matrix, which is generated by Position-Specific Iterated Basic Local Alignment Search Tool. Then, random forest–based classifiers are used to infer the protein interactions. When performed on PPI data sets of yeast, human, and Helicobacter pylori, we obtained good results with average accuracies of 92.10%, 97.03%, and 86.45%, respectively. To better evaluate the proposed method, we compared Gabor feature, Discrete Cosine Transform, and Local Phase Quantization. Our results show that the proposed method is both feasible and stable and the Gabor feature descriptor is reliable in extracting protein sequence information. Furthermore, additional experiments have been conducted to predict PPIs of other 4 species data sets. The promising results indicate that our proposed method is both powerful and robust.
Introduction
Proteins play significant roles in the life activities of cells and organisms, such as neurotransmission, DNA replication, and cycle control. Most of the diversity of cellular functions is based on protein-protein interactions (PPIs). Detecting PPIs is highly critical for the exploration of biological cellular mechanisms. With the advent of high-throughput techniques, such as mass spectrometric protein complex identification, 1 protein chip, 2 and yeast 2-hybrid system,3,4 considerable PPI data have been generated. However, high-throughput experiments are usually accompanied by high false positive and false negative rates and high cost. Moreover, these methods can hardly predict the whole PPI networks. 5 Under this situation, developing a novel computational method to predict unknown PPIs is more urgent than adopting the traditional experimental method to identify PPI.6,7
It is important to make full use of available PPI experimental data to develop computational methods. Many PPI databases, such as Human Protein References Database (HPRD), 8 Database of Interacting Protein (DIP), 9 and Molecular INTeraction database (MINT), 10 have been built after a number of experiments depicting PPI network. However, there are differences in protein structure information,11,12 protein domains, and so on. With new protein amino acid sequence data explosively growing, computational methods are urgently needed to detect the information of protein sequence.
In recent years, a number of computational methods have been proposed to extract the feature vectors mainly from the amino acid sequence.13 -16 The discriminative feature can improve the performance of a classification model, and some computational methods were based on Chou’s pseudo amino acid composition (PseAAC)17 -19 that retains the information of protein sequence, although it only considers the influence of 3 kinds of characteristics. Furthermore, some new methods on feature vector extraction are based on kernels. The method proposed by Jaakkola et al 20 is the first to use Fisher kernel to detect homology. Shen et al 21 proposed the support vector machine (SVM)-based method to predict PPIs. Leslie et al 22 put forward the mismatch string kernel method, which detects protein amino acid sequence at a lower computational cost. The difference between a PseAAC-based method and a kernel-based method lies in the way of extracting the feature information, with the first extracting the feature directly from the protein sequence and the second retaining some prior information and extracting feature vectors more effectively.
In general, most of the computational methods use machine learning algorithms combining various descriptors of proteins. Concerning different kinds of protein data, the main existing computational approaches can be divided into 2 categories: one uses information from the structure of proteins and genomic context; the other uses information from protein sequences. Moreover, newly discovered protein sequences grow exponentially in many different types of databases, and to shorten the gap between known protein sequence data and their interaction statuses, it is important to develop computational methods that directly use the information in protein sequences.
In this work, a novel computational method for predicting PPIs from an amino acid sequence based on a random forest (RF) 23 classification and a Gabor feature descriptor was proposed. The major improvement of this method is that it extracts protein sequence features through Gabor texture representation. Specifically, we adopted the Gabor feature representation on a Position-Specific Scoring Matrix (PSSM)24,25 to extract the evolutionary information from protein sequence, and then a classification RF is applied to infer the PPIs. In this way, each protein sequence is represented as a PSSM. To obtain more feature descriptors, we use a Gabor descriptor to extract features in each protein PSSM, and then each protein sequence is represented by 100-dimensional feature vectors. Two corresponding feature vectors would be joint together and represent a protein pair as a 200-dimensional feature vector. Finally, we used RF as a machine learning classifier for classification. The method was adopted for 3 PPI data sets from human, yeast, and Helicobacter pylori. Our results indicate that the computational method has good performance. To further evaluate the performance of this method, we compared the results of the proposed method with the support vector machine classifier to the Gabor feature, Discrete Cosine Transform (DCT), 26 and Local Phase Quantization (LPQ). 27 Moreover, we also used our approach for predicting the PPIs in 4 other species using the protein interaction data from yeast. The results of the proposed method in predicting PPIs indicate this approach is trustworthy.
Materials and Methods
Golden standard data sets
From the public DIP, we collected Saccharomyces cerevisiae PPI data sets. Then, the protein pairs that contain a protein with less than 50 residues or have more than 40% sequence identity were removed. The positive data set was constructed with the remaining 5594 protein pairs. The negative data set was constructed with the 5594 noninteracting protein pairs, which have different subcellular localization. Finally, we constructed 11 188 protein pairs, of which half are from the positive data set and half from the negative data set.
Two other PPI data sets were also collected. The first PPI data set was collected from the HPRD. The protein pairs with more than 25% sequence identity were removed. We constructed the golden standard positive data set with the remaining 3899 protein-protein pairs of experimentally verified PPIs from 2502 different human proteins. Following previous work, 28 we assumed that proteins in different subcellular compartments would not interact with each other. Therefore, 4262 protein pairs from 661 different human proteins were set as the golden negative data set. The complete human data set consists of 8161 protein pairs. The other PPI data set was constructed with 2916 H pylori protein pairs (1458 interacting pairs and 1458 noninteracting pairs), which were described by Martin et al. 29
Position-Specific Scoring Matrix
The PSSM is widely used in various biological research works, such as studies of subcellular localization, disordered regions, and protein secondary structure. The PSSM also has great potential in extracting evolutionary information from amino acid sequences. In this work, each protein sequence would be converted into PSSM by adopting a Position-Specific Iterated Basic Local Alignment Search Tool (PSI-BLAST). 24 The PSSM can be represented as follows:
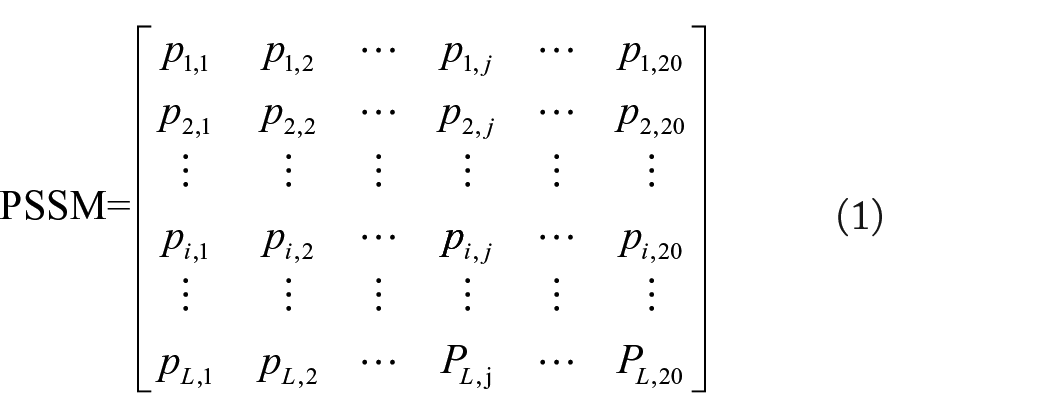
where L is the length of an amino acid sequence and Pi, j denotes the possibility that the ith amino acid of the given protein sequence mutates to amino acid j in the evolution process. To obtain highly homologous sequences, the parameters of PSI-BLAST (E-values) are set as 0.001 and 3 iterations are selected.
Gabor filter–based feature
First proposed by Gabor, 30 the Gabor filter is very similar to the visual stimulus response of cells in the human visual system. It has good characteristics in extracting local spatial and frequency domain information of targets. The Gabor feature is usually obtained by a convoluting image with a Gabor filter. Moreover, they have strong anti-interference ability in terms of image noise and illumination changes, and the most important advantages of Gabor filters are their translation, invariance to rotation, and scale. In image processing, the feature based on the Gabor filter is directly extracted from gray-level images. The 2-dimensional Gabor filter, in the spatial domain, is a Gaussian kernel function modulated by the complex sinusoidal plane wave. It can be defined as follows:
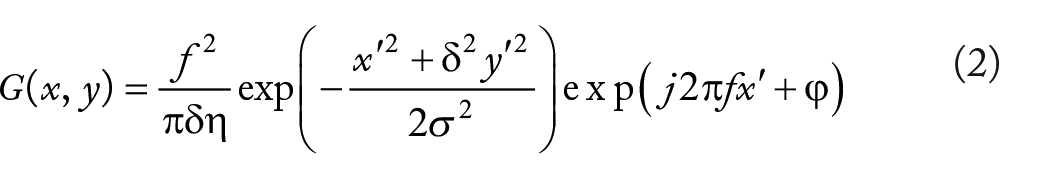


where
In our work, we use 40 Gabor filters in 5 scales and 8 orientations, which are shown in Figure 1. After using 40 Gabor filters, because of the high correlation of feature vectors, we can reduce the reduced feature data by way of downsampling for reducing information redundancy.31,32 Therefore, the protein sequence can be represented as Gabor feature vectors that are constructed by the first 100 coefficients.

Gabor filter in 5 scales and 8 orientations.
Random forrest classifier
At present, RF is one of the most popular prediction algorithms in data science. It was mainly developed by Breiman.
23
The RF model is one of the efficient ensemble classification algorithms, which uses multiple decision trees to reduce the output variance, thereby improving the accuracy of the classification. The RF classification makes full use of 2 powerful machine learning techniques. The first of RF classification is the selection of training examples, assuming that the original sample set has the total of examples
Results and Discussion
Evaluation measures
To better evaluate the proposed method, we calculated the following evaluation parameters: precision (PR), prediction accuracy (ACC), sensitivity (SN), specificity (SPC), and Matthew’s correlation coefficient (MCC). Their formation can be seen as follows:





where true negative (TN) is the number of true noninteracting pairs that are predicted correctly; true positive (TP) is the number of true samples that are predicted correctly; false positive (FP) is the number of true noninteracting pairs that are predicted to be interacting; and false negative (FN) is the correct number of samples that are predicted incorrectly. Moreover, the receiver operating characteristic (ROC) curves are one of the ways to evaluate the performance of the proposed method, and based on the prediction result, the area under an ROC curve (AUC) can also be computed to summarize ROC curve numerically.
Assessment of prediction ability
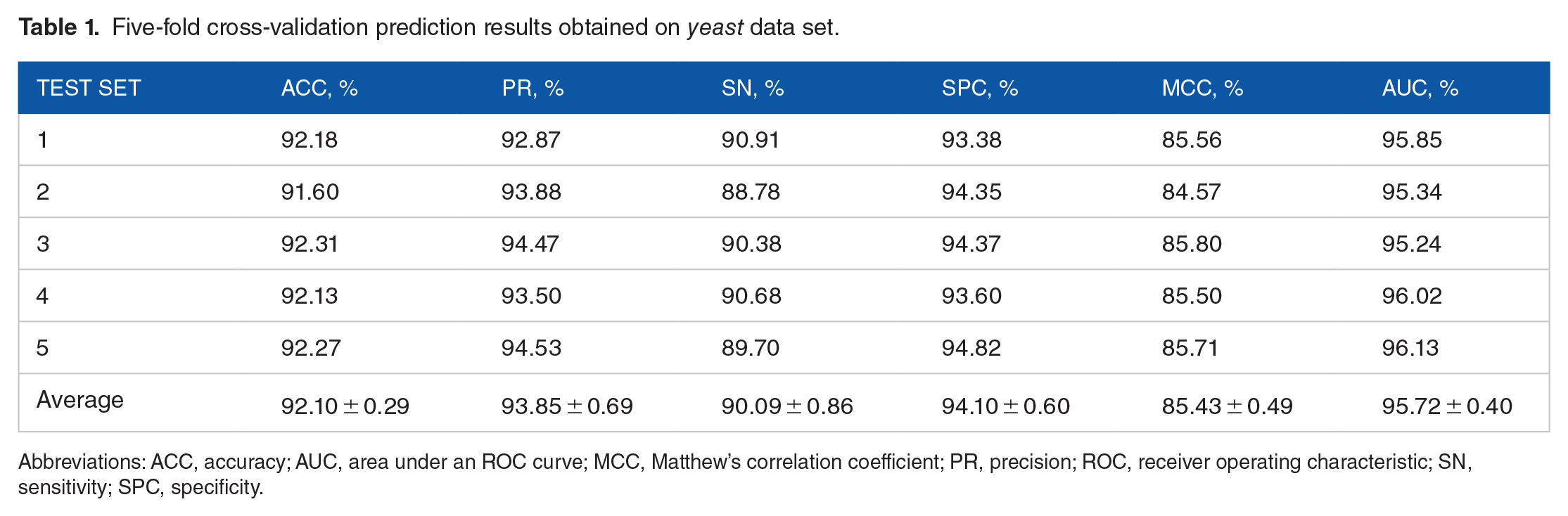
To ensure fairness of experiments, we conducted experiments in 3 different data sets of yeast, human, and Helicobacter pylori. We set the same corresponding parameters (N-tree = 100) in an RF classifier. Furthermore, to increase the credibility of our method, we adopted a 5-fold cross-validation to divide the whole data set into 5 parts, of which one-fifth is used for testing and four-fifths are used for training. By doing this, we can generate 5 models from the original data set. The prediction result based on RF classification models of protein sequence on 3 data sets is shown in Tables 1 to 3.
Five-fold cross-validation prediction results obtained on yeast data set.
Abbreviations: ACC, accuracy; AUC, area under an ROC curve; MCC, Matthew’s correlation coefficient; PR, precision; ROC, receiver operating characteristic; SN, sensitivity; SPC, specificity.
Five-fold cross-validation prediction results obtained on human data set.

Abbreviations: ACC, accuracy; AUC, area under an ROC curve; MCC, Matthew’s correlation coefficient; PR, precision; ROC, receiver operating characteristic; SN, sensitivity; SPC, specificity.
Five-fold cross-validation prediction results obtained on Helicobacter pylori data set.

Abbreviations: ACC, accuracy; AUC, area under a ROC curve; MCC, Matthew’s correlation coefficient; PR, precision; ROC, receiver operating characteristic; SN, sensitivity; SPC, specificity.
As shown in the tables, when predicting PPIs of yeast data set, the proposed approach can obtain prediction performance with an average accuracy, precision, sensitivity, specificity, and MCC of 92.10%, 93.85%, 90.09%, 94.10%, and 85.43% and standard deviations of 0.29%, 0.69%, 0.86%, 0.60%, and 0.49%, respectively. When predicting PPIs of human data set, the average accuracy, precision, sensitivity, specificity, and MCC are 97.03%, 98.01%, 95.74%, 98.22%, and 94.22% and standard deviations are 0.20%, 0.53%, 0.41%, 0.50%, and 0.36%, respectively. When predicting PPIs of Helicobacter pylori data set, the average accuracy, precision, sensitivity, specificity, and MCC are 86.45%, 88.51%, 83.82%, 89.06%, and 76.53% and standard deviations are 0.90%, 0.82%, 1.55%, 1.37%, and 1.30%, respectively. The ROC curves are shown in Figures 2 to 4. The y-ray depicts the true positive rate and the x-ray depicts the false positive rate in these figures. Meanwhile, the AUC values were also computed and the results of yeast, human, and Helicobacter pylori data sets were 95.72%, 99.33%, and 91.89%, respectively.

ROC curves performed by the proposed method on yeast protein-protein interaction data sets. AUC indicates area under an ROC curve; ROC, receiver operating characteristic.

ROC curves performed by the proposed method on human protein-protein interaction data sets. AUC indicates area under an ROC curve; ROC, receiver operating characteristic.

ROC curves performed by the proposed method on Helicobacter pylori protein-protein interaction data sets. AUC indicates area under an ROC curve; ROC, receiver operating characteristic.
According to these results, the method is both practical and effective for predicting PPIs by combining the Gabor feature with RF classification. Furthermore, these criterion values in low deviations indicate that the method we proposed is stable and robust. The main advantage of the feature extraction method is that it can not only retain enough prior information of PSSM but also describe the sequence information of protein sequence efficiently. The ability of the Gabor feature in obtaining effective information in PSSM is outstanding. Besides, considering the influence of protein sequence order, the texture information extracted by the Gabor feature can retain the effective information of protein sequence well. The results show that the utilization of the Gabor texture feature to extract evolutionary information in predicting PPIs in the proposed method is effective.
Comparison with other feature extraction methods
To evaluate the effectiveness of the Gabor feature in extracting protein sequence information and identifying protein interactions, we further compared the results to DCT and LPQ with the same RF classification. The DCT algorithm is a popular linear separable transformation. It is mainly used for data or image compression, and DCT has a good performance of decorrelation due to its ability to convert signals from the spatial domain to the frequency domain. The LPQ is considered as an effective operator for texture feature descriptors, which remain the blur-invariant property, and the LPQ is also widely used in facial recognition and image processing. In our work, the DCT feature, the LPQ feature, and the Gabor feature were extracted from PSSM, and then we made a comparison in the same RF classification.
The results of yeast and H pylori PPI data sets are presented in Figure 5. From Figure 5A to F, the Gabor feature basically dominates the LPQ feature and the DCT feature in terms of accuracy, specificity, sensitivity, precision, MCC, and AUC. In the yeast data set, the accuracy, specificity, and MCC gaps between Gabor and LPQ are 3.09%, 0.16%, and 4.45%, respectively. And the accuracy, specificity, and MCC gaps between Gabor and DCT are 0.50%, 0.23%, and 3.28%, respectively. Similarly, in the Helicobacter pylori data set, the accuracy, specificity, and MCC gaps between Gabor and LPQ are 6.22%, 4.35%, and 8.41%, respectively. And the accuracy, specificity, and MCC gaps between Gabor and DCT are 7.36%, 5.26%, and 9.73%, respectively. Thus, the Gabor feature has little difference in specificity from LPQ and DCT, but, due to the difference in specificity from MCC, we speculate that the Gabor feature has better performance than LPQ in extracting texture feature of protein sequence, especially in enhancing the sequence information of a protein sequence. The Gabor feature is similar or even better than DCT in extracting protein sequence information.

Performance comparison with 6 validation metrics using the Gabor feature (blue bar), the LPQ feature (green bar), and the DCT feature (yellow bar). (A) Accuracy rates, (B) sensitivity, (C) precision, (D) specificity, (E) MCC and (F) AUC. AUC indicates area under an ROC curve; DCT, discrete cosine transform; LPQ, local phase quantization; MCC, Matthew’s correlation coefficient; ROC, receiver operating characteristic.
Performance on the independent data sets
As we obtained good results on 3 PPI data sets of yeast, human, and Helicobacter pylori, for further evaluating the proposed method, we assumed that homologous proteins can preserve their ability to interact and used interactions experimentally identified in one organism to predict interactions in other organisms. The basis of this assumption is that homologs have similar functional behaviors. Therefore, they preserve the same PPI. 33 The 4 independent data sets we used share low identity with the training data set. Specifically, 11 188 samples from the yeast data set were used as the training data set. Then, we tested on 4 PPI data sets that were independent of the training data set. These data sets were treated as positive data sets that have been converted into PSSM. The experimental results of the 4 independent data sets are summarized in Table 4. The prediction performance accuracy was 93.20%, 94.89%, 91.93%, and 91.34% on Caenorhabditis elegans, Mus musculus, Homo sapiens, and Helicobacter pylori, respectively. It demonstrates that our proposed method can yield a superior prediction performance toward cross-species data sets.
Model prediction results of 4 species.

Abbreviation: ACC, accuracy.
According to the results in Table 4, when the PPI data set from yeast was used as the positive samples to predict the PPIs of other cross-species, the prediction performance was effective. And it is noteworthy that the prediction model is constructed using S cerevisiae PPI data set, so the trained model represented the characteristics of S cerevisiae PPI. Meanwhile, a strong generalization ability demonstrates that the method we proposed is effective. Moreover, it is reliable to assume that PPIs generated in one species can be used to predict PPIs in other species. The number of PPIs in one organism might have “coevolved” with another organism, so their corresponding orthologs interact as well. 34 This notion of conserved interactions is also supported by the observation that many interactions are conserved between different species in molecular machines or signal transduction pathways. 35
Comparison with other methods
In recent years, a large number of algorithms have emerged for predicting PPIs. In Table 5, we compared previous studies that proposed other methods to predict PPIs of Helicobacter pylori data set. The accuracy of other methods is between 75.80% and 87.50%; our proposed method is slightly lower than ensemble ELM (extreme learning machine) in accuracy, sensitivity, and MCC, so we assumed that ensemble ELM classification is more effective than RF classification in learning classification. We then compared our method with the existing methods of yeast and human data sets. The results of 6 other methods with accuracy ranging from 75.08% to 92.10% are shown in Table 6. The proposed method gets high average accuracy (92.10%), which is higher than other methods. The lower standard deviation also means that the performance of the proposed method is more robust. From Table 7, we can observe that the performance of our method is higher than that of previous work based on RF classification.
Performance comparison of different methods on the Helicobacter pylori data set.

Abbreviations: ACC, accuracy; ELM, extreme learning machine; HKNN, K-local hyperplane distance neighbor; MCC, Matthew’s correlation coefficient; PR, precision; SN, sensitivity.
Performance comparison of different methods on the yeast data set.

Abbreviations: ACC, accuracy; LD, local descriptor; MCC, Matthew’s correlation coefficient; PCA-EELM, principal component analysis-ensemble extreme learning machine; PR, precision; RF, random forest; SN, sensitivity; SVM, support vector machine.
Performance comparison of different methods on the human data set.

Abbreviations: ACC, accuracy; LDA, latent Dirichlet allocation; MCC, Matthew’s correlation coefficient; PR, precision; RF, random forest; RoF, rotation forest; SN, sensitivity; SVM, support vector machine.
From the table above, we can see that using an ensemble classifier such as the ensemble of HKNN (K-local hyperplane distance nearest neighbor) and boosting is better than using a single classifier, which can achieve more accurate and robust performance. Through these comparisons, and compared with the most advanced methods at present, we can observe that the RF-based model combined with PSSM can improve the prediction accuracy directly. Feature extraction containing evolutionary information and the selection of classifiers are the primary way to promotion. Meanwhile, its excellent performance demonstrates that the Gabor feature has a strong ability in extracting protein sequence information, especially enhancing protein texture features. Thus, effective feature extraction improves the performance of classification.
Conclusions and Discussion
In recent years, the number of researchers requiring more knowledge to detect PPIs is increasing. Due to the complexity and high dimensionality of proteomic data, flexible and powerful statistical learning tools are needed for effective statistical analysis, which promotes the rapid development of computing methods for predicting PPIs. In this article, we proposed a novel computational method for predicting PPIs in which an RF classifier combined with the Gabor feature descriptor on the PSSM is used. The main improvements of the proposed method are that the Gabor feature can extract the discriminative information of protein sequence, especially enhancing the texture feature information of protein sequence that the interaction between proteins is more likely to occur in the region with higher energy. The experimental results demonstrated that the good performance of our proposed method in predicting PPIs. The results also showed that Gabor features perform better than LPQ and DCT in texture feature and protein sequence correlation extraction. In future studies, more effective feature extraction methods and machine learning techniques will be explored for PPI prediction.
Footnotes
Author Contributions
XKZ and ZHY conceived and designed the analysis. ZHY and LPL provided data. YL, ZW and XZK provided mathematical theory and performed simulations. XZK and JP wrote the manuscript. All authors reviewed the final manuscript.
Declaration of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research is funded by the National Natural Science Foundation of China under Grant (No. 61722212 and 61873212.)