
Abstract
Influenza A has caused several deadly pandemics throughout human history. The virus is often resistant to developed treatments because of its genetic drift or shift property. Broad-spectrum antibodies show a promising potential to overcome the resistance of influenza viruses. In silico studies on broad-reactive antibodies and their interactions with hemagglutinins might shed light on the rational design of a universal vaccine. In this study, 11 broad-spectrum antibodies (or antigen-binding fragments) and 14 hemagglutinins of H3N2 and H5N1 strains were docked and analyzed to provide information about the construction of the scaffold for using universal antibodies against the influenza A virus. Antigen-binding fragments that have high number of appearances in the top 3 within each H3 and H5 subtypes were chosen for protein-protein interaction analysis. The results show that while the hydrogen bond is important for Ab/Fab binding to H3, the H5-Ab/Fab system may need cation-pi interaction for a strong interaction.
Introduction
Influenza A is one of the deadliest infectious diseases in human history, and it can be ranked equally with the bubonic plague in the ability to create large-scale fear and terror despite the great effort of humans to control it.1-3 From the first outbreak in 1918, called the “Spanish flu” and caused by the H1N1 strain, influenza A has killed 4 to 5 times more people than the number of casualties in World War I. In 1957, the Asian flu, caused by influenza A H2N2, began in China and spread around the world, killing 1.1 million people in total. 3 Ten years later, a new strain of H3N2 lead to the eruption of a new pandemic called the “Hong Kong flu” that caused the death of 1 million people. 3 In 2009, the strain that caused a deadly pandemic in World War I returned with a new type called H1N1pdm09; the result was approximately 575 000 deaths. 3 Along with the deadly strains that infect humans, other strains, like H5N1, cause problems that can indirectly and catastrophically effect humans. 4 The H5N1 outbreak appeared in Vietnam in 2003 when a massive avian infection led to the forcible killing of poultries, crippling the agriculture industry. 4
Influenza A disease is named after its causative agent, influenza A virus, a member of the Orthomyxoviridae family. It is distinguished by its structure of hemagglutinin (HA) and neuraminidase proteins. Hemagglutinin is a glycoprotein that appears in large numbers on the outer membrane of the virus. The trimeric HA0 comprises two types of subunits, with 3 HA1 making the globular head and 3 HA2 making the cylindrical tail. HA1 and HA2 have distinct roles in beginning and mediating the infection, respectively. 2 Initial infection occurs when the globular head binds to the sialic acid receptors on the host membrane and triggers the cellular endocytosis to kill the influenza A virus. After entering the cell, the lower pH of the endocytosic endosomes allows the cleavage of the cylindrical tail from the globular head. This action causes the fusion of the host endosome membrane and the viral membrane to occur, together with the release of viral RNA and proteins into the host cytoplasm. Meanwhile, neuraminidase (NA), a polypeptide containing approximately 470 amino acids, participates at the end of the infection cycle by preventing viral particles to be aggregated during the budding of the new virus. 2 After reassembling, the HAs of the virus are still attached to the sialic acids on the surface membrane of the host cells, and NA has to cleave an α-ketosidic linkage between HA and the sialic acid receptor on the host cells to release the virus and act as a posttranslational modification for HA. This virus can fight back against the immune system and treatments with great resistance by means of their segmented genome. 2 The genome is made of 8 segments, each encoded for one specific part of the virus.2,5 This unique structure not only allows the virus to mutate easily—called an antigenic shift—and evade the treatment, but it also allows 2 or more strains of virus to combine together and form a new type.2,5
In the modern medical battlefield, fighting against influenza A virus is crucial and intense. The threat of the next deadly influenza pandemic is constant, and some strains of seasonal flu can become lethal if patients do not receive proper and prompt attention. Approved treatment for influenza A in humans includes the usage of some chemicals, such as Oseltamivir (Tamiflu), Zanamivir (Relenza), and Peramivir (Rapivab). 6 Unfortunately, resistance to these drugs is beginning to be seen. Some mutations, such as H274Y or N294S on the H1N1pdm09 strain, or H274Y and N294S on the H5N1 strain, are resistant to Oseltamivir. 7 Three mutations of H1N1pdm09 have been observed after treatment with Zanamivir. While chemical reagents require time for direct action on the virus and can cause side effects in the user, antibodies create an immediate shield for the body. A monoclonal antibody injection can create temporary immunity against viruses at the beginning of the outbreak while more specific treatments are being developed. 8 However, the development of monoclonal antibodies against influenza A viruses faces many difficulties, such as low production and poor storage conditions, high expense, long research time, high risk of failure in clinical trials, and the potential for a quick antigenic drift of the virus. 5 In addition, the particular strain of influenza A virus that might arise cannot be predicted, and a monoclonal antibody can only recognize one type of protein. The broad-spectrum antibody possesses the ability to recognize multiple types of viruses, and it can still be effective against another strain if one strain is beginning to immune; thus, broad-spectrum antibodies can reduce the time and cost of developing new antibodies. The neutralizing action of an antibody against an antigen depends on the binding affinity between the receptor and its ligand. Four main interactions that contribute to binding are a hydrogen bond, a hydrophobic interaction, an ionic interaction, and a cation-pi interaction. Hydrophobic interaction is formed when proteins are surrounded by water or components that have hydrophobicity interact with each other to minimize the exposed surface. In a hydrogen bond, which is a strong dipole-dipole interaction, one atom always has hydrogen at the end while another atom contains an electronegative charge. The electrostatic charge density also affects the strength of the interaction, including in an ionic or cation-pi interaction. The ionic interaction occurs between two opposite charged groups—a positive charge side chain and a negative charge group—and a cation-pi interaction is the non-covalent interaction formed by a positive charge residue with an aromatic side chain.
In this research, an in silico approach was used on the systems of 11 broad-reactive antibodies (Abs) or antigen-binding fragments (Fabs) and 14 HAs from the H3 and H5 subtypes. The results were analyzed to determine the main interacting pattern between the HA and its neutralized Ab/Fab for use in contributing to the building of the scaffold of universal antibodies used against the influenza A virus. Because of the distinct role of HA and NA, HA is widely chosen as a subject for research on the use of antibodies against influenza.
Materials and Methods
Protein preparation
There are 64 Abs/Fabs that can actively neutralize HAs that have been published in the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) since 1998. Only 11 are considered broad-spectrum Abs/Fabs for their ability to bind and neutralize more than one subtype of HA (Table 1). Meanwhile, 114 859 HA proteins from different influenza A strains have been submitted to the UnitProt database since 1986. A total of 167 proteins have been reviewed by SwissProt, of which there are 52 H3 and 27 H5. As influenza A can develop a resistance to the treatment quickly due to its antigenic shift, and as there is still a high risk for an outbreak to turn into a pandemic, it is important to focus on the most recent data to provide enough data with which to intercept and curtail the next opportunistic outbreak of influenza A. For this research, we selected 14 HAs that were isolated in 2000 and published between January 1, 2014, and December 31, 2018; these H3 and H5 belong to H3N2 and H5N1 strains that are still potential threats to humans (Table 1). The PyMOL program was used to extract HA and Ab/Fab proteins separately from the original .pdb file and save that data into raw files. The duplicated and non-related chains were also deleted. SwissPDB was used to correct atoms on the protein file and GROMACS was used to minimize the energy of the proteins.
List of antibodies/antigen-binding fragments and hemagglutinins used for research.
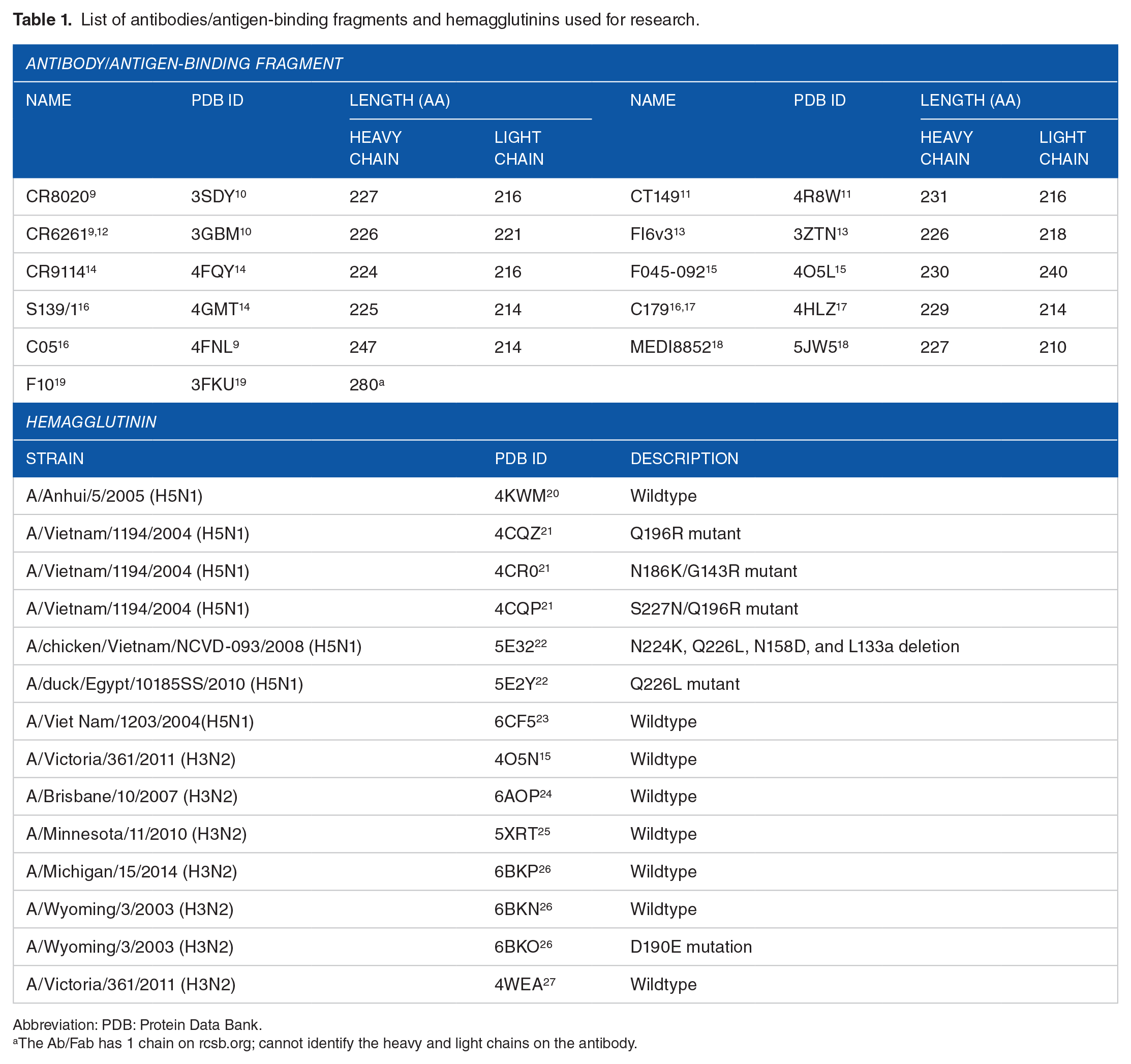
Abbreviation: PDB: Protein Data Bank.
The Ab/Fab has 1 chain on rcsb.org; cannot identify the heavy and light chains on the antibody.
Docking simulation
MEGADOCK 4.0 was used for the docking simulation between 11 Abs/Fabs and 14 HAs from the H3 and H5 subtypes. In this research, pairwise shape complementarity (rPSC), electrostatic, and desolvation free energy of receptor (RDE) models were chosen to calculate the docking score. The proteins were covered by 1.2 Å radius 3D voxels, which were scored differently depending on their position on the protein. 28 On the receptor (R), –45 is scored to the voxels if they were located in the receptor, while another score was calculated based on the number of atoms within a 3.6 Å radius of the Van der Waals radius if the voxel located on the open space. On the ligand (L), the score 1 was added for each voxel inside the protein, while 0 was given for the solvent accessible surface layer and the open space. A CHARMM19 force field was used to calculate the physicochemical score of the electrostatic interaction between amino acids, which, together with the electric field, is depended on the Euclidean distance between voxels. In the RDE model, a non-pairwise-type atomic contact energy (ACE) score of receptors was defined based on a table constructed according to the study of Zhang et al.29,30 The experiment was set with the number of rotational sampling angles at a maximum of 54 000, 2000 output predictions with 1 prediction per each rotation, no fast Fourier transform (FFT) point, and 1.0 as both the electrostatic and hydrophobic term ratio.
Protein-protein interaction analysis
Based on the difference in docking score, different combinations of Ab/Fab and HA were chosen for protein-protein interaction (PPI) analysis. The PPI analysis was performed by Protein Interactions Calculator (PIC), a web-based server from the Molecular Biophysics Unit, Indian Institute of Science, Bangalore. 31 The protein systems were examined to indicate all types of hydrogen bonds, hydrophobic interactions, ionic interactions, and cation-pi interactions. These interactions were calculated by MEGADOCK, as was previously used for the docking score. A pivot table was used to represent the involvement of key amino acids in different types of interaction between Ab/Fab and HA.
Statistical analysis
The highest docking score for each case was extracted for analysis. The docking scores of each Ab/Fab on each type of HAs were collected and analyzed using GraphPad Prism version 8. One-way analysis of variance (ANOVA) with the Geisser-Greenhouse correction was applied, together with Tukey multiple comparison tested on individual variances, to understand the difference between docking scores of Abs/Fabs on the H3 or H5 group. P-value was marked following APA style with significant difference at P-value ⩽0.05.
Results and Discussion
From the first outbreak of influenza, the urgency of finding a vaccine or treatment methods increases with the fear of a pandemic. Since 1998, there have been 64 promising Abs/Fabs that have neutralized effect on the HA of the influenza A virus. However, with the high frequency of antigen drift of this virus and the slow introduction of a vaccine, broad-spectrum Abs/Fabs are a priority. In this study, we examined the binding activity of different Abs/Fabs on the HAs H3 and H5 to understand the interacting patterns of broad-reactive Abs/Fabs.
A docking simulation was conducted between each Ab/Fab and each HA. The parameters were set at default and the docking scores were calculated based on rPSC, electrostatic, and RDE scores. In total, 154 cases were successfully docked under the provided conditions. The docking scores were collected and analyzed using 1-way ANOVA with pairwise comparison. Within one Ab/Fab, the docking scores on different HA were summarized and are represented in Figure 1. The observed results showed that, within 7 HAs of the H3 subtype, C05 has the highest docking score with a mean of 5602 and a standard deviation (SD) of 187. CR9114 and CR8020 take the second and third places with 5524 and 5467 in mean, respectively. This ranking does not coincide with the order made by the number of appearances in the top 3 within H3 proteins (Table 2). In contrast, CR9114 takes first place with 2 times, C05 is in second place, and third place is shared by Fabs F045-092 and MEDI8852. Meanwhile, within the 7 chosen proteins of the H5 subtype, MEDI8852 Ab takes the highest position in docking with 5241 and 337.4 in mean and SD, respectively. Second place belongs to FI6v3 and in third place is CR9114. This ranking also differs from that of the number of appearances in the top 3 within H5, in which FI6v3 keeps the first place, MEDI8852 takes the second place, and CR9114 takes the third together with C179 (Table 2). However, in the docking results, the ANOVA analysis showed an insignificant difference between the Abs/Fabs in the top 3 of each H3 or H5 subtype (Supplementary S01); thus, we used the top 3 Abs/Fabs ranked by number of appearances for further PPI analysis.

Docking score of 11 Abs/Fabs on hemagglutinins H3 and H5. The data are presented by average score with standard error of mean.
Number of appearance in the top 3 highest docking scores of Abs/Fabs within each type of H3 and H5.

To understand the difference in docking scores between the Ab/Fab–HA systems, further analysis of the PPI between CR9114, C05, F045-092, FI6v3, MEDI8852, or C179 on both H3 and H5 was conducted and compared. All kinds of hydrogen bonding, hydrophobic, ionic, and cation-pi interactions were calculated by the web-based server PIC. This analysis also gave the general pattern of interaction between the chosen broad-reactive Abs/Fabs with H3 and H5. Key amino acids which have frequent appearance in either one interaction or many interactions between Ab/Fab and HA are considered. It was observed that the number of amino acids of Abs/Fabs joining in interaction with H5 was less than the number of amino acids of Ab/Fabs joining in interaction with H3; also, the frequency of the appearance of these amino acids in a number of HAs within H5 was lower than within H3 (Figure 2). When doing a PPI analysis of CR9114 with the H3 subtype, there were no amino acids observed in the cation-pi interaction, which was the same result received when this Fab was analyzed with the H5 subtype. In addition, CR9114 had many amino acids with very high frequency in the hydrophilic interaction, although there was no multiple-interactive amino acid. The highest frequency found in the hydrophilic interaction belonged to SER168 and Leu170 with 85.7%—equal in appearance in 6 of 7 HAs. In contrast, C05 and F045-092 interacting with the H3 subtype showed no hydrogen bond formed by the system; a lot of amino acids were observed joining in the ionic interaction, while the cation-pi interaction had the least number of amino acids. The C05 antibody had 3 amino acids in the ionic interaction (ASP56, ILE51, and ILE57), 5 amino acids in the hydrophobic interaction (ILE51, ILE57, TYR79, TRP100, and VAL100), and 1 amino acid in the cation-pi interaction (TRP100) and was identified as having the highest frequency of appearance with 71.4%, which is equal to 5 of 7 HAs interactions. Specifically, 3 amino acids (ILE51, ILE57, and TRP100) not only contributed to the hydrophobic interaction but also were found two in the ionic interaction and one in the cation-pi interaction, respectively. With the large difference in number and frequency of amino acids, the hydrophobic interaction was considered the main interaction of C05 Fab with H3. In the system of F045-092 and H3, the amino acids LYS 13 and LYS156 were recorded in multiple interactions of the ionic and cation-pi action, while in the system formed by MEDI8852 and H3, there was no multiple interactions of amino acids. When we combined the docking result with the PPI analysis, it was seen that the hydrogen bond is more important than the other three in binding the Abs/Fab and the H3 subtype, especially in the case of C05 and F045-092, while the hydrogen bond has little role in the binding of these protein complexes. Hydrophobic interactions appear to act as additional strength for both F045-092 and C05 binding to H3, which makes the docking score of the C05 Fab on H3 higher than that of F045-092 and MEDI8852. While it is difficult to determine the main interaction between CR9114, C05, F045-092, and MEDI8852 with H3 proteins from many interacted amino acids, it is easy to observe the importance of the cation-pi interaction between the chosen Abs/Fabs and H5 proteins. For all 4 protein systems, only the first and second place Abs/Fabs were recorded having cation-pi interaction. FI6v3, which had the highest docking score and number of appearances in the top 3, was remarkable with a very small number of amino acids participating in forming interactions; however, these amino acids have a high frequency of appearance. Specifically, LYS43 is the only amino acid that joined in the cation-pi interaction with 71.4% in appearance frequency, 3 in hydrophobic interactions by VAL85 (57.1%), PHE139 (28.6%), and LEU166 (57.1%), and 4 amino acids in ionic interaction by ASP 9 (85.7%), ARG83 (28.6%), GLU85 (28.6%), and GLU161 (42.9%). The MEDI8852-H5 system is the only one among the formed Fab-HA systems having all 4 types of interaction; however, as in the frequency of appearance among the 7 proteins of the H5 subtypes, the interaction between this Fab on H5 was not preserved, which might give a different efficiency in different influenza A virus strain. The 2 third-place Abs within the H5 subtypes, CR9114 and C179, clearly showed no evidence of cation-pi interaction, together with a low frequency of the appearance of interacted amino acids.

Amino acids on 6 antibodies/antigen-binding fragments C179, CR9114, C05, F045-092, MEDI8852, and FI6v3 joined to the interaction with H3 and H5 show different frequency of contribution. The frequency is calculated based on times of appearance out of 7 H3 or 7 H5 hemagglutinins (with n/a: not available).
All 6 Abs/Fabs were analyzed not only in the group in which they were in the top 3 but also in the other group of HAs to find the common amino acids that appeared in both HA groups. These amino acids are considered key for broad-reactive Ab/Fabs and material for universal vaccine development. CR9114 has the common amino acids—PRO109, THR131, and SER168—for binding on both H3 and H5 subtypes. PRO109 joins in protein binding with a hydrophobic interaction, while THR131 and SER168 join in a hydrogen bond with both H3 and H5 subtypes. This Ab and MEDI8852 are the only two Abs that appeared in the top 3 of both chosen H3 and H5 proteins, despite the fact that there were no records of common amino acids for MEDI8852. The number of amino acids that met the requirement on the H5 subtype was larger than in the H3 subtype, but the maximum frequency was the same. In both the H3 and H5 subtypes, the maximum frequency was 57.14. SER65 was the only amino acid that formed a hydrogen bond in the H5 interactions that had the highest frequency, while there were 3 amino acids that had the same frequency in the H3 interactions, including TYR 32 in the hydrophobic interaction, VAL165 in the hydrogen bond, and LYS165 in the ionic interaction. The only recorded amino acid that contributed to multiple interactions was LYS145; this amino acid participated in forming a hydrogen bond and cation-pi interaction with a H5 protein. MEDI8852 is also the only antibody under clinical trial that was designed to neutralize all HA subtypes of influenza A virus. 18 In vitro observation concluded that MEDI8852 inhibits HA-mediated membrane fusion activity. The C05 antibody has a unique ability: not only does it have a wide range of neutralization, it can also neutralize the antibody with a very low binding affinity. 9 In a previous report, under in vivo conditions, a 10 mg/kg dose of C05 antibody protected mice from a lethal dose of the A/Aichi/2/X-31/1968 (H3N2) virus. 9 The PPI calculation shows that many of the amino acids from the C05 antibody can be considered key interactions due to a very high frequency; ILE51 and ILE57 both appeared in the hydrophobic and ionic interaction with a frequency of 71.43%. Only one amino acid, VAL100, showed a common appearance in both the H3 and H5 subtypes and acted as a hydrophobic linkage in both. F045-092 had a great effect on not only H3N2 but also on H1, H2, and H13 HA. 15 As it is binding, F045-092 uses its 23-residue HCDR3 to attack the binding site of the HA involved in receptor mimicry. 15 As we discovered, this Ab has 2 common amino acids acting in ionic interaction for binding with H3 and H5 subtypes (LYS 13 and GLU85). Ab C179 also has 4 common amino acids (ALA 11 , VAL 12 , SER 14 , and VAL84) that have a hydrophobic interaction with both subtypes. This Ab was previously found to recognize and neutralize the H1 and H2 subtypes of HA together; the docking stimulation shows the potential of this antibody for H5 subtype neutralization. 16 Despite the fact that the antibody has the most of docking scores are highest on H5 subtype, antibody FI6v3, which neutralized HAs in 1 to 10 subtypes in the enzyme-linked immunosorbent assay (ELISA) test, 13 contained very few amino acids, while the frequency was high. The highest frequency recorded was 85.71%, that equals to 6 of 7 HA interactions participated in, of amino acids ASP 9 joining in ionic interaction. This FI6v3 Ab has LYS43 and ARG83 as 2 common amino acids for both H3 and H5, which acted differently in the H3 and H5 interaction. If, in the H3 subtype, LYS43 acts in the ionic interaction, it serves as a cation-pi interaction in the H5 subtype, and vice versa with ARG83. It is not surprising that in all 12 systems of the 6 chosen Abs/Fabs and HAs, Leucine (LEU) had the most abundant amino acids, with Lysine (LYS) and Serine (SER) following. Leucine is found in both the hydrophobic interaction and the hydrogen bond. The structure of this amino acid includes one alkane area in opposition with the carboxyl and amine groups, allowing this amino acid to form both a hydrophobic interaction and a hydrogen bond. LYS is notable in its position as second in the appearance of common amino acids. The structure of Lysine is the reverse of Glutamic acid, with two amine groups and one carboxyl group, thus creating a positive charge to participate in the hydrogen bond and ionic interaction. In addition, Lysine was also found to have a cation-pi interaction in this research; this was due to the charge area of lysine interacting with a rich π system of protein. The structure of Serine contains 2 carboxyl groups and 1 amine group, which creates charges for this amino acid.
In this study, Ab CR9114 was determined to be the best broad-reactive Ab, not only because the docking score in the H3 subtype was superior but also because the PPI analysis showed a very high frequency of appearance of amino acids. Along with CR9114, MEDI8852 also deserves our attention; it not only appeared in both the top 3 in appearance of highest docking score, but also the average docking scores on H3 and H5 are similar. However, PPI analysis of this Fab shows interactive amino acids with quite low frequency and no common amino acid. This suggests that MEDI8852 may have a different effect on different strains. Thus, CR9114 has the most potential as an Ab for research on a universal vaccine against the influenza virus. This Ab can protect animals from a lethal dose of H3N2 with a 5 mg/kg injection. 14 The difference between docking scores shows that CR9114 has greater effect on the H3 subtype than on the H5. The docking score on the H5 subtype of this Ab is not as high as in the other top 3 Abs within the H5 subtype even though this Ab can bind to H5 HA. 16 C05 is in the top 3 of the Abs/Fabs having high docking scores with both H3 and H5 subtypes. However, the interaction between C05 and the H5 HA is not realistic. There is research that shows that the steric clash on the paratope of C05 prevents the interaction between this antibody and H5 HA. 32 The difference in this result and the results of the research by Wu et al could be explained by the fact that MEGADOCK cannot perform molecular dynamics, which is important in in vitro and in vivo experiments. This C05 result was then used as motivation for further research where the removal of steric clash is observed.
In the end, from the docking score and PPI analysis of 6 chosen Abs/Fabs—CR9114, C05, F045-092, C179, MEDI8852, and FI6v3—on 14 different structures of H3 and H5 proteins, it can be seen that while a hydrogen bond is important for Ab/Fab binding to H3, cation-pi interaction plays a role in the binding of Ab/Fab to H5. This can be extended to examine the effect of these interactions on different strains of the influenza virus to gather more information, and in vitro research can be used to confirm the results of the replacement of these amino acids.
Conclusions
Our docking experiments have shown the difference between antibodies when they interact with the same HAs. The result reveal that on the H3 subtype, CR9114, C05, F045-092, and MEDI8852 have the number of times their docking scores are highest, while on the H5 subtype, the same top 3 positions belong to FI6v3, MEDI8852, CR9914, and C179. The PPI analysis in H3 shows that CR9114 and C05 contain many amino acids with a high frequency of participation in interactions with H3 proteins, especially in the hydrogen bond. Meanwhile, the analysis of H5 interaction shows that cation-pi interaction is more important in the binding score of Abs/Fabs in the protein. Based on this docking simulation, CR9114 and MEDI8852 are good enough to be chosen for both H3 and H5 interaction. Our study reveals motif bindings and a common feature in these popular antibodies that is a significant component for the rational design of a universal vaccine. More molecular dynamic simulation should be conducted to observe key interactions with protein flexibility.
Supplemental Material
Supplementary_S01_xyz25376bd1938b9 – Supplemental material for Molecular Docking of Broad-Spectrum Antibodies on Hemagglutinins of Influenza A Virus
Supplemental material, Supplementary_S01_xyz25376bd1938b9 for Molecular Docking of Broad-Spectrum Antibodies on Hemagglutinins of Influenza A Virus by Khanh PB Le, Phuc-Chau Do, Rommie E Amaro and Ly Le in Evolutionary Bioinformatics
Footnotes
Funding:
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was funded by the Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 108.06-2017.332.
Declaration Of Conflicting Interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions
KPBL, P-CD and LL conceived and designed the experiments. KPBL and P-CD performed the experiments and analyzed the data. KPBL, P-CD, REA and LL wrote the paper.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.