To better understand the transmission and evolution of Mycobacterium tuberculosis (MTB) in Taiwan, six different MTB isolates (representatives of the Beijing ancient sublineage, Beijing modern sublineage, Haarlem, East-African Indian, T1, and Latin-American Mediterranean (LAM)) were characterized and their genomes were sequenced. Discriminating among large sequence polymorphisms (LSPs) that occur once versus those that occur repeatedly in a genomic region may help to elucidate the biological roles of LSPs and to identify the useful phylogenetic relationships. In contrast to our previous LSP-based phylogeny, the sequencing data allowed us to determine actual genetic distances and to define precisely the phylogenetic relationships between the main lineages of the MTB complex. Comparative genomics analyses revealed more nonsynonymous substitutions than synonymous changes in the coding sequences. Furthermore, MTB isolate M7, a LAM-3 clinical strain isolated from a patient of Taiwanese aboriginal origin, is closely related to F11 (LAM), an epidemic tuberculosis strain isolated in the Western Cape of South Africa. The PE/PPE protein family showed a higher dn/ds ratio compared to that for all protein-coding genes. Finally, we found Haarlem-3 and LAM-3 isolates to be circulating in the aboriginal community in Taiwan, suggesting that they may have originated with post-Columbus Europeans. Taken together, our results revealed an interesting association with historical migrations of different ethnic populations, thus providing a good model to explore the global evolution and spread of MTB.
Mycobacterium tuberculosis (MTB) continues to be a leading cause of human deaths by an infectious agent. It has been estimated that approximately one-third of the world's population has been infected with the tubercle bacillus, and 1.5 million die from MTB infection every year.1 In 2012, 12,338 new cases were reported in Taiwan, with an estimated annual incidence of 53 cases per hundred thousand people.2 Epidemiologic studies have revealed that different genotypes of MTB may be prevalent in different geographic regions worldwide and that genotype distribution is closely associated with population migrations.3–5 The distribution of human MTB genotypes is closely associated with geography, ethnicity, age, and host factors. Recent developments in DNA sequencing technologies have revolutionized tuberculosis (TB) research, contributing to major advances in understanding the evolution and pathogenesis of MTB and facilitating the development of new diagnostic tests with increased specificity for TB. Identification of the genomic features of major MTB strains is key to deciphering the transmission of virulence and drug resistance among different strains. Thus, comparative analysis of whole-genome sequences can provide better insights into the evolution of the MTB strains present in Taiwan. Achieving these goals will improve our understanding of the epidemiology of TB in Taiwan and help guide prevention policy.
Taiwan, a relatively isolated island in the southeast of mainland China, is regarded as a mixing vessel of immigrants over the past four centuries as colonization by different waves of ethnic groups occurred. The aborigines in Taiwan are of Austronesian descent, which distinguishes them from the major ethnic group on the island, the Han Chinese, and now reside predominantly in the mountainous regions or rural areas.6 Documentation of aboriginals on the island can be traced back to the 16th century, when Spanish sailors arrived and named the island. There are 12 aboriginal tribes on the island, which are presumed to represent the ancestral colonies that inhabited the island for at least 4000 years. The other two predominant ethnic populations of Taiwan are descended from Han Chinese who migrated to the island in two major waves: the first during the Ming Dynasty around 1600 and the second between 1945 and 1950, when members of the military, veterans, and some civilians emigrated from mainland China due to the civil war there7; in total, about two million mainland Chinese have migrated to Taiwan to date. Taiwan was occupied by the Dutch for 40 years beginning 1660, and the Japanese from 1895 until 1945.
We previously demonstrated that the Beijing ancient strain and the Haarlem strain are the predominant MTB strains infecting aborigines in eastern Taiwan (Hualien City), the East-African Indian (EAI) strain is prevalent in southern Taiwan aborigines, and the Beijing modern strain is predominant in Han Chinese.7–10 In the present study, six MTB strains – isolates of the Beijing ancient sublineage, the Beijing modern sublineage, Haarlem, EAI, T1, and Latin-American Mediterranean (LAM) – representing the major types of clinical strains isolated from three different ethnic groups (aboriginals, Han Chinese, “veterans”) in Taiwan7,8 were subjected to whole-genome sequencing.11–13 The six Taiwan genomes were then compared to four reference MTB strains (H37Rv, H37Ra, CDC 1551 (LAM), and F11 (LAM)14–16) as well as the genome of Mycobacterium bovis. The presence of significant sequence diversity in MTB could provide a basis for understanding pathogenesis, immune mechanisms, and bacterial evolution. Polymorphic genes are good candidates for virulence and immune determinants, because proteins that interact directly with the host are known to have elevated divergence. Examples in MTB are the PE/PPE genes that encode proteins with proline-glutamate and proline–proline–glutamate motifs.17
Methods
Study patients and bacterial isolates
Aborigines and veterans are entitled to government-subsidized health benefits under the National Health Insurance Claim System of Taiwan; therefore, the identities of these patients were confirmed by the type of insurance policy or the type of identification card. We obtained MTB isolates from three hospitals in three different regions of Taiwan. Patients with symptoms compatible with pulmonary TB and with sputum cultures positive for M. tuberculosis complex were included. Isolates were stored frozen by the participating hospitals and sent to the TB laboratory in the Division of Infectious Diseases of National Health Research Institutes (NHRI). MTB genomic DNA was extracted from primary LJ egg cultures as described previously.18 In this study, we first characterized the phenotypes and genotypes of at least 1000 isolates of MTB from different ethnic populations, at least 100 isolates from each population (aborigine, veterans, and Han Chinese), followed by a comparative genomics study to provide a snapshot of mycobacterial evolution and its pathogenesis.
Genotyping
All isolates were subjected to spoligotyping (ST) and determination of the 19 variable number of tandem repeats (VNTR)-mycobacterial interspersed repetitive unit (MIRU) loci. ST was performed as previously described by Kamerbeek et al.19 Data were compared with the international SpolDB4.0 database.20 VNTR-MIRU loci (ETR-A, B, C, D, E, F; MPTR-A; MIRU-2, 4, 10, 16, 20, 23, 24, 26, 27, 31, 39, 40) were individually amplified and analyzed as previously described by Supply et al.21 Results from each of the 19 loci were combined to create 19-digit allelic profiles.
Genome sequencing and DNA analysis
MTB strains were sequenced separately to 10- to 30-fold coverage of the genome using a Genome Sequencer 20 (GS20) or a Genome Sequencer FLX (GS FLX) instrument (454 Life Sciences, Roche)22 with a 500-800-base pair shotgun library for each strain. The reads generated by 454 pyrosequencing were assembled using the GS De Novo Assembler version 2.5.3 provided by the manufacturer. Protein-coding genes were predicted from all contigs of each assembly using GLIMMER version 3.02.23 All contig sequences of the six strains were compared to the reference strain H37Rv to detect single-nucleotide polymorphisms (SNPs) using MUMmer version 3.20.24 The genome sequence of H37Rv was downloaded from the NCBI ftp (ftp://ftp.ncbi.nih.gov/genomes/Bacteria/Mycobacterium_tuberculosis_H37Rv_uid57777/). MUMmer provides a pipeline with the programs “nucmer”, “delta-filter”, and “show-snps” for sequence aligning, repeat filtering, and SNP/INDEL calling. The pipeline is able to detect all SNPs between two genomes, even where there are many sequence rearrangements. Phylogenies based on the whole genomes of the six MTB strains sequenced in this study and the five completely sequenced strains available through NCBI were constructed using the MUMmer distance matrix method adopted by Chan et al.25 In brief, MUMs (maximal unique matches between the two genomes) obtained from MUMmer were summarized for each pair of genome sequences and then divided by the smaller genome length of the pair.
Construction of phylogenetic trees
The genetic diversity (based on MIRU-VNTR loci and ST) of the six selected Taiwan MTB strains was used to build a neighbor-joining tree. A pairwise distance can be obtained after log transformation and multiplication by -1. After the distance matrix of all the 11strains (the six Taiwan isolates and five reference strains) was completed, the program MEGA4.1 was applied to build a phylogenic tree using the minimum-evolution method (Fig. 1C).
Genetic diversity of six Taiwan MTB strains. Neighbor-joining tree showing genetic diversity of six Taiwan MTB strains based on MIRU-VNTR loci (A) and spoligotyping (B). A whole-genome sequencing-based dendrogram was generated using minimum evolution methods (C).
Screening of SNPs in the MTB genome
Although 454 sequencing allows rapid determination of the genome sequence, the data remain incomplete, with gaps and potential errors. To circumvent the time-consuming process of finishing, we bypassed the task of whole-genome alignment and instead used the reference strains CDC1551 and H37Rv as a framework against which to align each of our contigs separately by BLAST. The gene names, SNPs, and their locations in the contigs were all identified by this alternative procedure. Using the H37Rv genomic sequence as the reference, SNPs in the six Taiwan MTB strains were identified using MUMmer, and N substitutions and frame shifts were identified using BLAT.26 Genes encoding PE and PPE family proteins were identified using BLASTX. The number of S and N substitutions identified in PE/PPE family protein-coding genes and all other protein-coding genes for each of the strains (using H37Rv as the reference) are summarized in Table 1.
SNP identification in strains A18, A27, M3, M7, M24, and W6 using H37Rv as the reference.
PPE
ALL
SNPs
S
N
N/S
dn/ds
SNPs
S
N
N/S
dn/ds
EAI2_Manilla
A18
75
21
54
2.57
0.86
1777
681
1096
1.61
0.56
T1
A27
44
17
27
1.59
0.50
784
324
460
1.42
0.50
Haarlem-3
M3
50
20
30
1.50
0.49
743
306
437
1.43
0.50
LAM3
M7
35
12
23
1.92
0.60
762
307
455
1.48
0.52
Ancient Beijing
M24
78
23
55
2.39
0.82
1242
489
753
1.54
0.55
Modern Beijing
W6
60
30
30
1
0.35
1127
445
682
1.53
0.54
Avg.
57
21
37
1.83
0.60
1073
425
647
1.50
0.53
Statistical analyses
BioNumerics software (v 3.0; Applied Maths) was used to analyze MIRU by character types. Similarities between MIRU types were calculated using the categorical coefficient, in which all MIRU loci were weighted equally. This procedure counted the number of matched loci between pairs of isolates; when there was a difference, they were scored as unmatched, irrespective of the number of repeats present (thus, 1 versus 3 scored the same, ie, unmatched, as 1 versus 4). A dendrogram was constructed by the unweighted pair group method using arithmetic means averages. MIRU types were discriminated by similarity index and compared with octal format, ST, and family results.
Ethics statement
This study was approved by the Human Ethics Committee of the National Health Research Institutes, Taiwan (Code: EC0961103). Because of the retrospective nature, routine collection of clinical data in daily practice, and dislinkage of personal information, the requirement to obtain informed consent was waived by our institutional review board.
Results
Sequencing of six M. tuberculosis genomes from patients in Taiwan
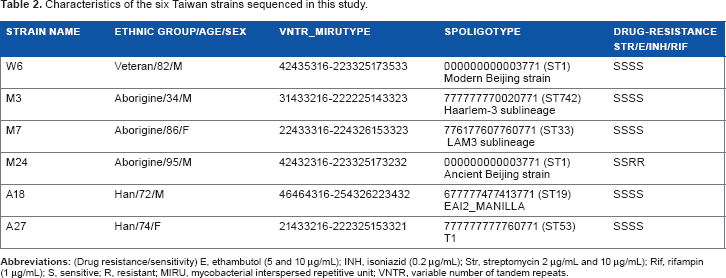
The characteristics (VNTR-MIRU type, spoligotype, and drug resistance/sensitivity) of the six MTB strains (M3: Haarlem-3; M7: LAM-3, W6: Beijing modern; A18: EAI; A27: T1; and M24: Beijing ancient) chosen for whole-genome shotgun sequencing are listed in Table 2.
Characteristics of the six Taiwan strains sequenced in this study.
STRAIN NAME
ETHNIC GROUP/AGE/SEX
VNTR_MIRUTYPE
SPOLIGOTYPE
DRUG-RESISTANCE STR/E/INH/RIF
W6
Veteran/82/M
42435316-223325173533
000000000003771 (ST1) Modern Beijing strain
SSSS
M3
Aborigine/34/M
31433216-222225143323
777777770020771 (ST742) Haarlem-3 sublineage
SSSS
M7
Aborigine/86/F
22433316-224326153323
776177607760771 (ST33) LAM3 sublineage
SSSS
M24
Aborigine/95/M
42432316-223325173232
000000000003771 (ST1) Ancient Beijing strain
SSRR
A18
Han/72/M
46464316-254326223432
677777477413771 (ST19) EAI2_MANILLA
SSSS
A27
Han/74/F
21433216-222325153321
777777777760771 (ST53) T1
SSSS
Abbreviations: (Drug resistance/sensitivity) E, ethambutol (5 and 10 μg/mL); INH, isoniazid (0.2 μg/mL); Str, streptomycin 2 μg/mL and 10 μg/mL); Rif, rifampin (1 μg/mL); S, sensitive; R, resistant; MIRU, mycobacterial interspersed repetitive unit; VNTR, variable number of tandem repeats.
Sequencing of the first three strains (M3, M7, and W6) was carried out using the GS20 pyrosequencing system, and the remaining three genomes (A18, A27, and M24) were sequenced using the GS FLX system, which doubled the read length from ~100 bases to more than 200 bases (Table 3; total number of contigs/bases; predicted number of coding genes). Initial reference mapping of the shotgun reads was performed using the complete genomic sequence of the highly studied MTB strain H37Rv (Table 3; number of SNPs; number of INDELs). Coverage ranged from about 10 × to 30 × with the GS20 system, and we achieved an assembly rate of over 96%, with the total assembled genomes ranging in size from 4.2 to 4.3 Mbp. This level of coverage significantly lengthened the average contig sizes and also reduced the number of large contigs (>800 nucleotide flows, about 500 called bases).
Genome assemblies of six Taiwan M. tuberculosis strains.
STRAIN ID (GENOTYPE)
TOTAL NO. OF CONTIGS
TOTAL NO. OF BASES
PREDICTED NO. OF CODING GENES
NO. OF SNPs
NO. OF INDELs
W6 (Modern Beijing)
507
4,311,575
4,236
1,605
215
M24 (Ancient Beijing)
396
4,246,251
4,279
1,544
164
M3 (Haarlem3)
442
4,316,882
4,219
981
119
M7 (LAM3)
539
4,304,264
4,275
918
185
A27 (T1)
364
4,273,576
4,389
932
104
A18 (EAI2)
380
4,286,824
4,443
2,161
279
Note: The SNP and INDEL results were obtained using the MUMmer package by comparing all contig sequences of each strain to the H37Rv reference sequence.
Pairwise comparison of the M. tuberculosis genomes
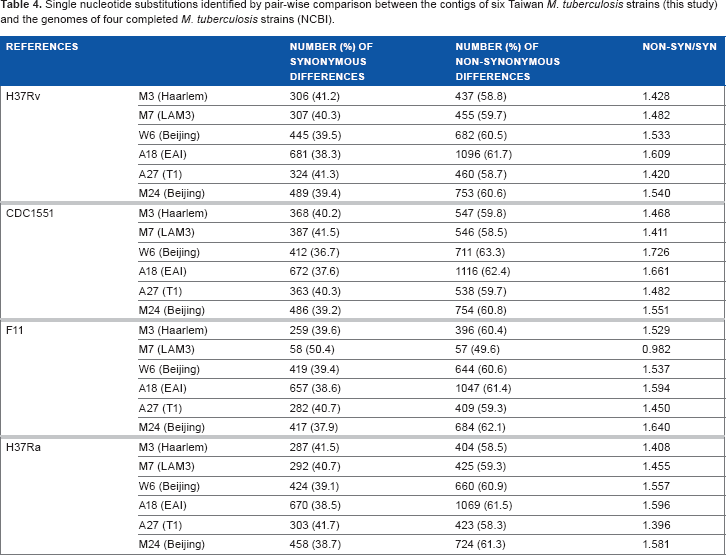
The coding sequences of each of the six Taiwan MTB strains were compared against the completely sequenced genomes of five MTB strains available through NCBI: H37Rv, H37Ra, CDC1551, F11, and KZN. The number of SNPs identified in the coding sequences of M3, M7, and A27 were lower compared to the other three strains when H37Rv was used as the reference strain, suggesting that the former three are more closely related to H37Rv (Table 3). Notably, the number of SNPs identified in the coding sequences of M3, M7, and A27 were lower than the number of SNPs identified in the coding sequences of W6, M24, and A18. In all pairwise comparisons, the percentages of nonsynonymous (N) substitutions ranged from 58% to 63%, except in the case of M7 and F11. The M7 strain, a LAM-3 sublineage collected from a Taiwanese aboriginal patient, showed a strikingly lower number of single-nucleotide substitutions when compared against the Western Cape F11 strain, isolated from a TB epidemic in South Africa (Table 4), suggesting a closer relatedness. The number of synonymous (S) and N nucleotide substitutions identified between M7 and F11 are nearly equal (S = 58; N = 57).
Single nucleotide substitutions identified by pair-wise comparison between the contigs of six Taiwan M. tuberculosis strains (this study) and the genomes of four completed M. tuberculosis strains (NCBI).
REFERENCES
NUMBER (%) OF SYNONYMOUS DIFFERENCES
NUMBER (%) OF NON-SYNONYMOUS DIFFERENCES
NON-SYN/SYN
H37RV
M3 (Haarlem)
306 (41.2)
437 (58.8)
1.428
M7 (LAM3)
307 (40.3)
455 (59.7)
1.482
W6 (Beijing)
445 (39.5)
682 (60.5)
1.533
A18 (EAI)
681 (38.3)
1096 (61.7)
1.609
A27 (T1)
324 (41.3)
460 (58.7)
1.420
M24 (Beijing)
489 (39.4)
753 (60.6)
1.540
CDC1551
M3 (Haarlem)
368 (40.2)
547 (59.8)
1.468
M7 (LAM3)
387 (41.5)
546 (58.5)
1.411
W6 (Beijing)
412 (36.7)
711 (63.3)
1.726
A18 (EAI)
672 (37.6)
1116 (62.4)
1.661
A27 (T1)
363 (40.3)
538 (59.7)
1.482
M24 (Beijing)
486 (39.2)
754 (60.8)
1.551
F11
M3 (Haarlem)
259 (39.6)
396 (60.4)
1.529
M7 (LAM3)
58 (50.4)
57 (49.6)
0.982
W6 (Beijing)
419 (39.4)
644 (60.6)
1.537
A18 (EAI)
657 (38.6)
1047 (61.4)
1.594
A27 (T1)
282 (40.7)
409 (59.3)
1.450
M24 (Beijing)
417 (37.9)
684 (62.1)
1.640
H37Ra
M3 (Haarlem)
287 (41.5)
404 (58.5)
1.408
M7 (LAM3)
292 (40.7)
425 (59.3)
1.455
W6 (Beijing)
424 (39.1)
660 (60.9)
1.557
A18 (EAI)
670 (38.5)
1069 (61.5)
1.596
A27 (T1)
303 (41.7)
423 (58.3)
1.396
M24 (Beijing)
458 (38.7)
724 (61.3)
1.581
Phylogeny of MTB strains based on genomic sequencing and VNTR-based genotyping
The six Taiwan MTB strains and the five reference strains were included in this analysis (Fig. 1). This phylogenetic tree (Fig. 1B) is almost congruent with the one we constructed using the neighbor-joining method as well as with the 17 loci MIRU-based analysis (Fig. 1A).
SNP identification in strains A18, A27, M3, M7, M24, and W6 using H37Rv as the reference
The six strains have similar N/S ratios when all SNPs in the protein-coding genes are taken into consideration. It is notable that three of the strains (A27, M7, and M3) have fewer SNPs and may be evolutionarily more closely related to H37Rv. The genes for the PE/PPE family proteins in A27, M3, and M7 also have fewer SNPs in comparison to the other strains. The dn/ds ratio for the PE/PPE family was higher in A18 (EAI2_Manilla strain) compared to the average dn/ds ratio for the PE/PPE family.
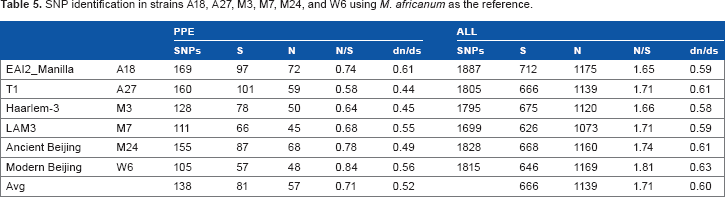
SNP identification in strains A18, A27, M3, M7, M24, and W6 using Mycobacterium africanum as the reference
SNP identification was also performed for the six strains using the M. africanum strain GM041182 (NC_015758) as the reference. The results are similar to those using H37Rv as the reference (Table 5). In contrast to the SNPs for all protein-coding genes, most of the strains showed slightly lower dn/ds values in the PE/PPE family protein genes, except A18.
SNP identification in strains A18, A27, M3, M7, M24, and W6 using M. africanum as the reference.
PPE
ALL
SNPs
S
N
N/S
dn/ds
SNPs
S
N
N/S
dn/ds
EAI2_Manilla
A18
169
97
72
0.74
0.61
1887
712
1175
1.65
0.59
T1
A27
160
101
59
0.58
0.44
1805
666
1139
1.71
0.61
Haarlem-3
M3
128
78
50
0.64
0.45
1795
675
1120
1.66
0.58
LAM3
M7
111
66
45
0.68
0.55
1699
626
1073
1.71
0.59
Ancient Beijing
M24
155
87
68
0.78
0.49
1828
668
1160
1.74
0.61
Modern Beijing
W6
105
57
48
0.84
0.56
1815
646
1169
1.81
0.63
Avg
138
81
57
0.71
0.52
666
1139
1.71
0.60
SNP identification in strains A18, A27, M3, M7, M24, and W6 using Mycobacterium cannettii and Mycobacterium marinum as references
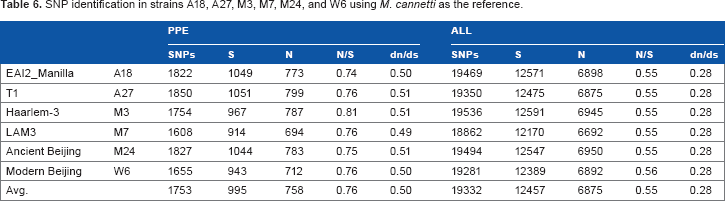
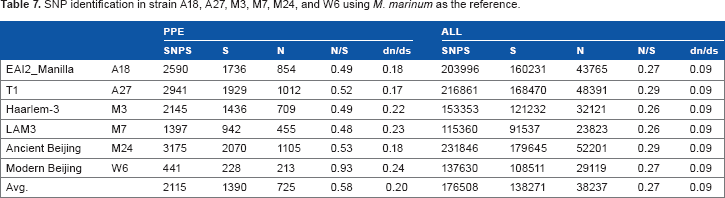
SNP identification was performed for the six strains using M. cannettii (NC_015848) as the reference (Table 6) and also M. marinum (accession no. CP000854) as the reference, a near relative of MTB with a 6.63-Mb genome containing 5424 coding sequences (Table 7). Comparison of the six Taiwan strains to M. cannettii or M. marinum yielded many more nucleotide substitutions compared to using H37Rv or M. africanum as the reference. However, the dn/ds ratio was found to be much lower here.
SNP identification in strains A18, A27, M3, M7, M24, and W6 using M. cannetti as the reference.
PPE
ALL
SNPs
S
N
N/S
dn/ds
SNPs
S
N
N/S
dn/ds
EAI2_Manilla
A18
1822
1049
773
0.74
0.50
19469
12571
6898
0.55
0.28
T1
A27
1850
1051
799
0.76
0.51
19350
12475
6875
0.55
0.28
Haarlem-3
M3
1754
967
787
0.81
0.51
19536
12591
6945
0.55
0.28
LAM3
M7
1608
914
694
0.76
0.49
18862
12170
6692
0.55
0.28
Ancient Beijing
M24
1827
1044
783
0.75
0.51
19494
12547
6950
0.55
0.28
Modern Beijing
W6
1655
943
712
0.76
0.50
19281
12389
6892
0.56
0.28
Avg.
1753
995
758
0.76
0.50
19332
12457
6875
0.55
0.28
SNP identification in strain A18, A27, M3, M7, M24, and W6 using M. marinum as the reference.
PRE
ALL
SNPS
S
N
N/S
dn/ds
SNPS
S
N
N/S
dn/ds
EAI2_Manilla
A18
2590
1736
854
0.49
0.18
203996
160231
43765
0.27
0.09
T1
A27
2941
1929
1012
0.52
0.17
216861
168470
48391
0.29
0.09
Haarlem-3
M3
2145
1436
709
0.49
0.22
153353
121232
32121
0.26
0.09
LAM3
M7
1397
942
455
0.48
0.23
115360
91537
23823
0.26
0.09
Ancient Beijing
M24
3175
2070
1105
0.53
0.18
231846
179645
52201
0.29
0.09
Modern Beijing
W6
441
228
213
0.93
0.24
137630
108511
29119
0.27
0.09
Avg.
2115
1390
725
0.58
0.20
176508
138271
38237
0.27
0.09
Analysis of M. bovis genomic SNPs using M. marinum as the reference
We extracted the sequence data for the 3953 coding sequences from the M. bovis genome (accession no. BX248333) and mapped them to the M. marinum genome. In total, 231,941 SNPs were identified in protein-coding genes and, among these, 49,900 are nonsynonymous substitutions. The N/S ratio was determined to be 0.27 for all protein-coding genes. A total of 1104 of the SNPs belong to PE/PPE family protein genes, of which 315 are nonsynonymous substitutions. The N/S ratio for PE/PPE family protein genes was determined to be 0.399. For the PE/PPE family or all genes, the N/S ratio for the SNPs identified in M. bovis using M. marinum as the reference is <1. This is very different from the previous analysis for the six Taiwan MTB strains against M. marinum, in which the SNPs of the PE/PPE family have a higher N/S ratio than that of all genes (average 0.58 for PE/PPE versus 0.27 for all).
Discussion
Comparison of nonsynonymous substitutions per nonsynonymous site to the number of synonymous substitutions per synonymous site (dn/ds) between homologous genes is an important index in molecular evolution. In this study, we compared the dn/ds ratios among the SNPs called from mapping the sequencing reads of six MTB strains to different reference genomes.17 There are two types of natural selection in biological evolution: (1) positive selection promotes the spread of beneficial alleles and (2) negative selection hinders the spread of deleterious alleles.27 In our results, the average dn/ds values for SNPs called using H37Rv or M. africanum were found to be much higher than those using M. cannettii and M. marinum as references. Apparently, higher dn/ds ratios in genes encoding the PE/PPE proteins occur when genomes of relatively distantly related species such as M. cannettii and M. marinum are used as the reference genome (Tables 6 and 7). In other words, different selection effects are applied to PE/PPE protein genes versus other protein-coding genes in Mycobacterium evolution. Our observation coincides with those of previous studies that suggested a general positive selection or relaxation of negative selection in the molecular evolution of PE/PPE proteins in MTB.28 We believe that the second explanation is more likely in the case of MTB for the following reasons. (a) The effective population size of MTB has been significantly reduced because of its pathogenic lifecycle. This reduction in population size usually leads to decreased efficiency of selection, thus allowing deleterious mutations to accumulate in the genome. (b) The MTB–M. marinum dn/ds ratios are generally smaller than the ratios between different MTB strains. This is analogous to “smaller between-species distance than within-species distance.” The small (between-species) divergence usually indicates negative selection, whereas the larger (within-species) diversity usually means relaxation of negative selection, unless some type of diversifying selection has been in action.
Diversifying selection can cause remarkable genetic and phenotypic differences between different strains. One good example is the skin color of different human races. However, we will need stronger evidence and good biological explanations to claim this type of selection in MTB. In addition, we would like to emphasize that relaxed negative selection can sometimes be the source of functional innovations. A classical theory is “evolution by duplication,” which means that duplicated genes (as in the case of the PPE family) are subject to relaxed negative selection because of functional redundancy. Therefore, they are free to evolve and may accidentally acquire new functions that may increase the fitness of the carrier organisms. Such functions and the related genes will be subject to positive selection afterward. So the type of selection actually changes over time and with the context of evolution. In contrast, the differing dn/ds ratios between PE/PPE protein genes and all protein-coding genes are not detectable in most of the Taiwan MTB strains when H37Rv or M. africanum is used as the reference. The A18 MTB strain (EAI2_Manilla) is an exception, in that it was the only strain that showed elevated dn/ds values in the PE/PPE family, regardless of the reference genome used for SNP calling. This suggests a different fate for PE/PPE proteins in the evolution of the A18 strain in contrast to the other five strains from Taiwan.
Due to a complex interaction between the host, the pathogen, and the environment, the outcome of MTB infection and disease is highly variable.29,30 There is mounting evidence that this variable outcome may be influenced by MTB genomic diversity.29,31 However, the evolutionary forces that shape this variation are not well understood. Genomic comparisons have identified genetic variation for population screening; however, these analyses are limited to relatively few genetic loci that vary between the compared genomes and therefore are potentially misleading.14,32,33 Nucleotide sequences provide robust data for studying population variation. The mutational processes that generate this variation are understood, and sequence data have been successfully used in the study of bacterial epidemiology, population structure, and evolution.34 The complete genome sequences14–16,34 provide access to all regions of the chromosome and facilitate such studies.
A previous study of MTB strains in Taiwan by our group (based on ST and VNTR-MIRU analysis) revealed an interesting association of strains with historical migrations of different ethnic populations.35 Comparing whole-genome sequences of the main MTB strains in Taiwan in the present study confirmed the previous findings, thus establishing a good model to explore the global evolution and spread of MTB. The genome sequences of MTB strains isolated from representatives of Taiwanese aborigines (M3/Haarlem-3, M7/LAM-3, M24/Beijing ancient strain), a representative of a recent Han immigrant (W6/Beijing modern strain), and representatives of historic Han immigrants (A18/EAI2_MANILLA, A27/T1) were determined by 454 sequencing technology.11 More than 95% of the reads were assembled into sequence contigs. The sequence data from these representative strains will be further analyzed to discover the unique genomic features of MTB infecting different ethnic groups in Taiwan.
At present, we are focusing on MTB genomic information together with epidemiological and clinical data in order to identify factors significant in transmission, virulence, drug resistance, and protection efficacy of vaccines among different strains. We conducted Ka/Ks analysis to identify selection pressure on the protein-coding regions. As shown in Table 7, we found that, in general, there are more N than S changes in the MTB-coding sequences. Second, we found that the M7 strain isolated from an aboriginal patient is most closely related to the F11, an MTB strain isolated from a TB epidemic in the Western Cape of South Africa. This finding further supports our previous study, in which we demonstrated that the Haarlem and LAM lineages were circulating in the aboriginal community in Taiwan and suggesting a link of these strains to post-Columbus Europeans.6,36
The Beijing strains have spread worldwide as a genetically conserved genotype of MTB, often in association with drug resistance.8,37–40 This worldwide spreading in the population structure of MTB is driven in part by man-made factors, and perhaps also linked with intrinsic mycobacterial characteristics. By using DNA MassARRAY® technology (Sequenom), we have established protocols for rapid and cost-effective assays for distinguishing different sublineages of Beijing strains.33 Moreover, we found SNPs in a putative DNA repair gene, which may be involved in facilitating spreading of the pathogen, but did not demonstrate an association with multidrug resistance.33,41–45 Furthermore, we are conducting informatics analysis of the six sequenced Taiwan MTB genomes. Preliminary data indicate as many as 620 SNPs in at least two of the sequenced strains. The information generated by comparative analysis is the basis for establishing an MTB genotyping procedure for tracing the evolution and distribution of different MTBs in Taiwan. Molecular population genetic analysis of clinical strains delineates relationships among closely related strains of pathogenic microbes and allows construction of genetic frameworks for examining the distribution of biomedically relevant traits, such as virulence, transmissibility, and host range. In seeking to describe the distribution of characteristics of MTB strains and identify the determinants of that distribution, we are attempting to identify factors that determine disease transmission. Comparative genomic hybridization microarray chips will be designed based on the determined genomic sequences in order to conduct population genetic studies quickly and efficiently. Such studies will not only help us to understand the dynamics of TB transmission in Taiwan but will also combine sequence analysis and microarray technology for investigating drug resistance and virulence.
Conclusions
We demonstrated that Haarlem and LAM MTB strains are present in the aboriginal community in Taiwan, suggesting a link of these strains to post-Columbus Europeans. Taken together, our results revealed an interesting association of MTB strains with historical migrations of different ethnic populations, thus providing a good model to explore the global evolution and spread of MTB.
Author Contributions
Conceived and designed the experiments: H-YD, S-FT. Analyzed the data: Y-YC, Y-TC, J-RC, C-HL, K-MW, M-SL. Wrote the first draft of the manuscript: H-YD, Y-YC, Y-TC, C-HL, I-JS, S-FT. Contributed to the writing of the manuscript: H-YD, Y-YC, Y-TC, C-HL, S-FT. Made critical revisions and approved the final version: H-YD, Y-YC, Y-TC, J-RC, C-HL, K-MW, M-SL, I-JS, S-FT. All the authors reviewed and approved the final manuscript.
Footnotes
Acknowledgments
We thank the mycobacteriology laboratories of Mennonite Christian Hospital, Tri-Service General Hospital, and Wan-Ciao Veterans Hospital for providing bacterial isolates. All participants of this consortium are acknowledged for valuable discussions.
References
1.
World Health Organization.Goal Tuberculosis Control: Surveillance, Planning, Financing.Geneva: World Health Organization; 2008.
2.
Center for Disease Control.Tuberculosis Control Report 2013.Taipei: Center for Disease Control, Department of Health; 2013.
3.
TsolakiA.G., HirshA.E., DeRiemerK.. Functional and evolutionary genomics of Mycobacterium tuberculosis: insights from genomic deletions in 100 strains. Proc Natl Acad Sci U S A.2004; 101(14): 4865–70.
4.
HirshA.E., TsolakiA.G., DeRiemerK., FeldmanM.W., SmallP.M.Stable association between strains of Mycobacterium tuberculosis and their human host populations. Proc Natl Acad Sci U S A.2004; 101(14): 4871–6.
5.
GagneuxS., DeRiemerK., VanT.. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A.2006; 103(8): 2869–73.
6.
TrejautJ.A., KivisildT., LooJ.H.. Traces of archaic mitochondrial lineages persist in Austronesian-speaking Formosan populations. PLoS Biol.2005; 3(8): e247.
7.
DouH.Y., TsengF.C., LuJ.J.. Associations of Mycobacterium tuberculosis genotypes with different ethnic and migratory populations in Taiwan. Infect Genet Evol.2008; 8(3): 323–30.
8.
DouH.Y., TsengF.C., LinC.W.. Molecular epidemiology and evolutionary genetics of Mycobacterium tuberculosis in Taipei. BMC Infect Dis.2008; 8: 170.
9.
ChenY.Y., ChangJ.R., HuangW.F.. Molecular epidemiology of Mycobacterium tuberculosis in aboriginal peoples of Taiwan, 2006-2011. J Infect.2014; 68(4): 332–7.
10.
DouH.Y., ChenY.Y., KouS.C., SuI.J.Prevalence of Mycobacterium tuberculosis strain genotypes in Taiwan reveals a close link to ethnic and population migration. J Formos Med Assoc.2015; 114(6): 484–8.
11.
LiaoY.C., LiuT.T., ChangJ.R.. Draft genome sequence of Mycobacterium tuberculosis clinical strain W06, a prevalent Beijing genotype isolated in Taiwan. Genome Announc.2015; 3(6): e1460–15.
12.
LiaoY.C., ChenY.Y., LinH.H.. Draft genome sequence of the Mycobacterium tuberculosis clinical isolate C2, belonging to the Latin American-Mediterranean family. Genome Announc.2014; 2(3): e536–14.
13.
LiaoY.C., ChenY.Y., LinH.H.. Draft genome sequences of the Mycobacterium tuberculosis clinical strains A2 and A4, isolated from a relapse patient in Taiwan. Genome Announc.2014; 2(5): e672–14.
14.
FleischmannR.D., AllandD., EisenJ.A.. Whole-genome comparison of Mycobacterium tuberculosis clinical and laboratory strains. J Bacteriol.2002; 184(19): 5479–90.
15.
ColeS.T., BroschR., ParkhillJ.. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature.1998; 393(6685): 537–44.
16.
GarnierT., EiglmeierK., CamusJ.C.. The complete genome sequence of Mycobacterium bovis. Proc Natl Acad Sci U S A.2003; 100(13): 7877–82.
17.
FishbeinS., van WykN., WarrenR.M., SampsonS.L.Phylogeny to function: PE/PPE protein evolution and impact on Mycobacterium tuberculosis pathogenicity. Mol Microbiol.2015; 96(5): 901–16.
18.
van SoolingenD., de HaasP.E., HermansP.W., van EmbdenJ.D.DNA fingerprinting of Mycobacterium tuberculosis. Methods Enzymol.1994; 235: 196–205.
19.
KamerbeekJ., SchoulsL., KolkA.. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol.1997; 35(4): 907–14.
20.
BrudeyK., DriscollJ.R., RigoutsL.. Mycobacterium tuberculosis complex genetic diversity: mining the fourth international spoligotyping database (SpolDB4) for classification, population genetics and epidemiology. BMC Microbiol.2006; 6: 23.
21.
SupplyP., AllixC., LesjeanS.. Proposal for standardization of optimized mycobacterial interspersed repetitive unit-variable-number tandem repeat typing of Mycobacterium tuberculosis. J Clin Microbiol.2006; 44(12): 4498–510.
KurtzS., PhillippyA., DelcherA.L.. Versatile and open software for comparing large genomes. Genome Biol.2004; 5(2): R12.
25.
ChanP., LamT., YiuS., LiuC.A more accurate and efficient whole genome phylogeny. In: TaoJ., ed. In Proceedings of 4th Asia-Pacific Bioinformatics Conference.Taipei: Imperial College Press, London; 2005: 337–52.
26.
KentW.J.BLAT – the BLAST-like alignment tool. Genome Res.2002; 12(4): 656–64.
27.
PageR.D.M., HomlmesE.C.Molecular Evolution: A Phylogenetic Approach.Oxford: Blackwell Science; 1998.
28.
RileyR., PellegriniM., EisenbergD.Identifying cognate binding pairs among a large set of paralogs: the case of PE/PPE proteins of Mycobacterium tuberculosis. PLoS Comput Biol.2008; 4(9): e1000174.
29.
CawsM., ThwaitesG., DunstanS.. The influence of host and bacterial genotype on the development of disseminated disease with Mycobacterium tuberculosis. PLoS Pathog.2008; 4(3): e1000034.
30.
ComasI., GagneuxS.The past and future of tuberculosis research. PLoS Pathog.2009; 5(10): e1000600.
31.
KongY., CaveM.D., ZhangL.. Association between Mycobacterium tuberculosis Beijing/W lineage strain infection and extrathoracic tuberculosis: insights from epidemiologic and clinical characterization of the three principal genetic groups of M. tuberculosis clinical isolates. J Clin Microbiol.2007; 45(2): 409–14.
32.
BroschR., GordonS.V., MarmiesseM.. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci U S A.2002; 99(6): 3684–9.
33.
GutackerM.M., SmootJ.C., MigliaccioC.A.. Genome-wide analysis of synonymous single nucleotide polymorphisms in Mycobacterium tuberculosis complex organisms: resolution of genetic relationships among closely related microbial strains. Genetics.2002; 162(4): 1533–43.
34.
MaidenM.C., BygravesJ.A., FeilE.. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A.1998; 95(6): 3140–5.
35.
DouH.Y., HuangS.C., SuI.J.Prevalence of Mycobacterium tuberculosis in Taiwan: a model for strain evolution linked to population migration. Int J Evol Biol.2011; 2011: 937434.
36.
ChuangP.C., LiuH., SolaC., ChenY.M., JouR.Spoligotypes of Mycobacterium tuberculosis isolates of a high tuberculosis burden aboriginal township in Taiwan. Infect Genet Evol.2008; 8(5): 553–7.
37.
ChangJ.R., LinC.H., TsaiS.F.. Genotypic analysis of genes associated with transmission and drug resistance in the Beijing lineage of Mycobacterium tuberculosis. Clin Microbiol Infect.2011; 17(9): 1391–6.
38.
GlynnJ.R., WhiteleyJ., BifaniP.J., KremerK., van SoolingenD.Worldwide occurrence of Beijing/W strains of Mycobacterium tuberculosis: a systematic review. Emerg Infect Dis.2002; 8(8): 843–9.
39.
FilliolI., DriscollJ.R., van SoolingenD.. Snapshot of moving and expanding clones of Mycobacterium tuberculosis and their global distribution assessed by spoligotyping in an international study. J Clin Microbiol.2003; 41(5): 1963–70.
40.
DrobniewskiF., BalabanovaY., NikolayevskyV.. Drug-resistant tuberculosis, clinical virulence, and the dominance of the Beijing strain family in Russia. JAMA.2005; 293(22): 2726–31.
41.
AlmeidaD., RodriguesC., AshavaidT.F., LalvaniA., UdwadiaZ.F., MehtaA.High incidence of the Beijing genotype among multidrug-resistant isolates of Mycobacterium tuberculosis in a tertiary care center in Mumbai, India. Clin Infect Dis.2005; 40(6): 881–6.
42.
ToungoussovaO.S., CaugantD.A., SandvenP., MariandyshevA.O., BjuneG.Impact of drug resistance on fitness of Mycobacterium tuberculosis strains of the W-Beijing genotype. FEMS Immunol Med Microbiol.2004; 42(3): 281–90.
43.
ParkY.K., ShinS., RyuS.. Comparison of drug resistance genotypes between Beijing and non-Beijing family strains of Mycobacterium tuberculosis in Korea. J Microbiol Methods.2005; 63(2): 165–72.
IwamotoT., YoshidaS., SuzukiK., WadaT.Population structure analysis of the Mycobacterium tuberculosis Beijing family indicates an association between certain sublineages and multidrug resistance. Antimicrob Agents Chemother.2008; 52(10): 3805–9.