
Abstract
Deadenylases catalyze the shortening of the poly(A) tail at the messenger ribonucleic acid (mRNA) 3′-end in eukaryotes. Therefore, these enzymes influence mRNA decay, and constitute a major emerging group of promising anti-cancer pharmacological targets. Herein, we conducted full phylogenetic analyses of the deadenylase homologs in all available genomes in an effort to investigate evolutionary relationships between the deadenylase families and to identify invariant residues, which probably play key roles in the function of deadenylation across species. Our study includes both major Asp-Glu-Asp-Asp (DEDD) and exonuclease-endonuclease-phospatase (EEP) deadenylase superfamilies. The phylogenetic analysis has provided us with important information regarding conserved and invariant deadenylase amino acids across species. Knowledge of the phylogenetic properties and evolution of the domain of deadenylases provides the foundation for the targeted drug design in the pharmaceutical industry and modern exonuclease anti-cancer scientific research.
Introduction
Shortening of the polyadenylated (poly(A)) tail at the mRNA 3′-end, referred to as deadenylation, is a key step in mRNA decay in eukaryotes.1,2 This process is catalyzed by the deadenylase enzymes. Poly(A) tails are the preferred substrates of deadenylases, although in some instances they are capable of degrading non-adenosine ribopolymers in vitro with reduced efficiency.3–6 According to Goldstrohm and Wickens, 7 the known deadenylases are classified into 2 superfamilies, DEDD and exonuclease-endonuclease-phospatase (EEP), which are defined by conserved exonuclease sequence motifs required for catalysis. Members of the EEP superfamily of deadenylases use a conserved glutamic acid (E) and a histidine (H) for catalysis.7,8 This superfamily includes the families carbon catabolite repressor 4 (CCR4), Nocturinin, ANGEL and 2′ phosphodiesterase (2′PDE). 7 The DEDD superfamily of deadenylases is named after the invariant catalytic acidic residues aspartic acid (D) and glutamic acid (E), which are distributed in 3 exonuclease motifs.7,9 The DEDD superfamiliy includes the families POP2, Poly(A)-specific ribonuclease (PARN), CAF1Z and PAB-dependent poly(A)-specific ribonuclease subunit 2 (PAN2).7,9 In the present study, we focus on the molecular evolution of these families, thus providing insights into the amino acid conservation patterns that may be subsequently used for further studying deadenylases as a promising and emerging anti-cancer pharmacological target.
Methods
Identification of deadenylase homologues
To identify homologous deadenylase protein sequences, the accession numbers of the characterized deadenylases reported in literature 7 were used to retrieve their corresponding amino acid sequences from publicallyavailable databases UniProtKB 10 and GenBank. 11 These sequences were subsequently used as probes to search the sequence databases by applying reciprocal BLASTp and tBLASTn. 12 This process was reiterated until no new putative deadenylase homologues could be found.
Motifs construction
Representative DEDD and EEP peptide sequences were aligned and edited with Utopia suite's CINEMA alignment editor. 13 Sequence motifs were collected from the alignments, manually edited for insertions or gaps, and submitted to WebLogo3 14 to generate consensus sequences.
Phylogenetic analyses
The deadenylase sequences under study were searched against InterPro 15 in order to identify the boundaries of the core nuclease domain. In order to optimize the alignment and avoid unreasonable gap penalties, the amino acid sequences that correspond to the nuclease domain were collected from the entire deadenylase peptide sequences and aligned using CLUSTALW. 16 The resulting multiple sequence alignment was first trimmed for gaps using Gblocks17,18 and manually edited. The trimmed alignment was then used to reconstruct phylogenetic trees by employing 2 different methods. The first one is the maximum-likelihood method implemented in PhyML, 19 where an initial distance-based tree (BIONJ) is optimized using a hillclimbing algorithm. In our study, the nearest-neighbor-interchange (NNI) heuristic was used with 4 substitution-rate categories; the proportion of the invariable sites and the gamma shape parameter were estimated from the data. The number of amino acid substitutions per site was estimated with the LG 20 model. The second one is the Neighbor-net method 21 implemented in SplitsTree4, 22 a distance-based method which detects conflicting phylogenetic signals, presented in the form of reticulations; the Uncorrected P substitution model was used. Bootstrap analyses (1000 pseudo-replicates) were conducted in order to assess the robustness of the reconstructed trees. The inferred phylogenetic trees were visualized with the program Dendroscope. 23
Evolutionary rate shift analysis
A maximum-likelihood method 24 was employed for the identification of evolutionary rate differences at specific protein sites in DEDD families. Towards this end, a set of 19 protein sequences from the four DEDD families was analyzed in order to identify amino acid positions with significant 4 rate differences among the DEDD families as described in Knudsen and Miyamoto (2001). 24 The alignment was based on the core nuclease domain, and it was carried out using CLUSTALW. 16
Results/Discussion
Phylogenetic analyses of deadenylases
In the present study, we performed comprehensive and updated phylogenetic analyses of the deadenylase homologs in all available genomes (Figs. 1, 2, S1 and S2, Table S1). Collectively, 114 DEDD and 97 EEP homologous protein sequences were identified in the genomes of 38 and 37 species, respectively, which represent major eukaryotic taxonomic divisions (according to the NCBI taxonomy database; Table S1). 25
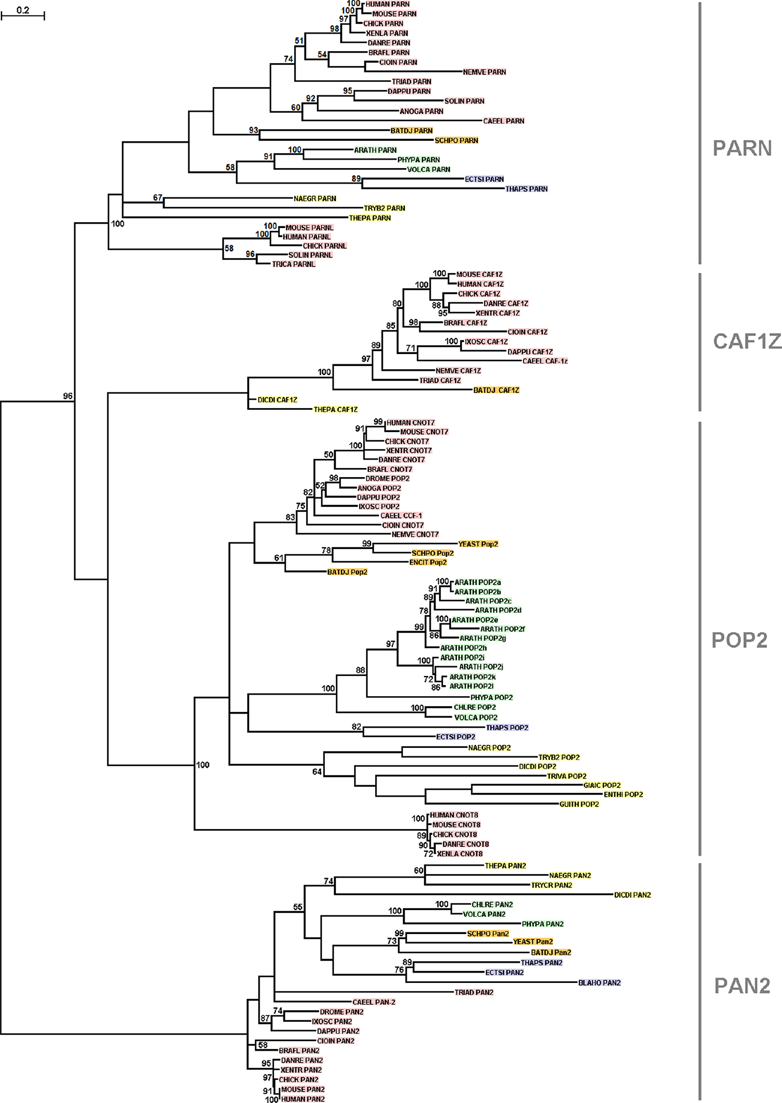
Phylogenetic tree of DEDD deadenylases. Bootstrap values (>50%) are shown at the nodes. The length of the tree branches reflects evolutionary distance. The scale bar at the upper left represents the length of amino acid substitutions per position. To minimize confusion, we used the protein names as described in Goldstrohm and Wickens; 7 the UniProt 5-letter codes were used for the species names. The proteins derived from metazoa are shown in red, from viridiplantae in green, from fungi in orange and from protozoa in yellow.

Phylogenetic tree of EEP deadenylases. Bootstrap values above 50% are shown at the nodes. The length of the tree branches depicts evolutionary distance. The scale bar at the upper left represents the length of amino acid substitutions per site. To minimize confusion, we used the protein names as described in Goldstrohm and Wickens; 7 the UniProt 5-letter codes were used for the species names. The proteins derived from metazoa are shown in red, from viridiplantae in green, from fungi in orange and from protozoa in yellow.
In order to better resolve the evolutionary relationships between the deadenylase families, we applied 2 different methods for phylogenetic tree reconstruction. The phylogenetic trees reconstructed with both methods are congruent, since the overall topology is similar, and all main branches are supported by high bootstrap values (Figs. 1, 2, S1 and S2). 8 coherent, well-supported monophyletic branches that correspond to the 4 families of the DEDD superfamily (Figs. 1 and S1), and the 4 families that comprise the EEP superfamily (Figs. 2 and S2) are distinguished.
Based on our analysis, putative members of the families POP2, PARN, PAN2 and CCR4 were identified in the major eukaryotic taxonomic divisions, ranging from metazoa to protozoa. POP2 appears to be the largest family in size with a wide distribution among taxa (Figs. 1 and S1).
Based on the phylogenetic analyses (Figs. 1, 2, S1 and S2), the deadenylase families POP2, PARN and CCR4 appear to have undergone gene duplications in metazoa giving rise to the, metazoan-specific, subfamilies CNOT8, PARNL and CNOT6 L, respectively. In POP2 and CCR4 families, gene duplications have rather occurred after the emergence of teleosts (bony fishes) (Figs. 1, S1, 2 and S2), since teleost (DANRE) homologs were detected in the corresponding subfamilies CNOT8 and CNOT6L. In PARN, a duplication event has presumably followed the radiation of arthropods (Figs. 1 and S1), as arthropod (SOLIN and TRICA) homologs were identified in the PARNL subfamily. However, neither frog (XENTR) nor fish (DANRE) PARNL homologs were identified; we suggest that frog and fish
Of importance, PARN homologs were not detected in the fungus
However, the deadenylase families CCR4-associated factor 1Z (CAF1Z), ANGEL, Nocturnin and 2′PDE are restricted to certain eukaryotic taxa (Figs. 1, S1, 2 and S2). CAF1Z is restricted to metazoa and protozoa (Figs. 1 and S1). Moreover, a putative CAF1Z homolog was detected in the chytrid fungus
Based on the rate shift analysis, a total of 153 sites, distributed across the core domain, were detected with significant evolutionary rate differences. Among them, 29 (19%) sites were detected with significant rate shifts between the PAN2 family and the other DEDD families (Fig. 3, both blue and red highlighting). This is in agreement with the phylogenetic analyses results where PAN2 appears to be more distantly related to the other DEDD families. Moreover, 29 (19%) conserved sites were detected in all DEDD families, exhibiting slower evolutionary rates compared to the average of all proteins under investigation (Fig. 3, blue highlighting). This leads to the suggestion that must have been evolutionary pressure to these sites to evolve slowly because they have a critical role in the function or structure of DEDD enzymes; as expected, the 4 catalytic residues that define the DEDD super-family are also included in this category (Fig. 3). Also, 95 sites, a significantly high percentage (62%), were detected with faster evolutionary rates compared to the average of all DEDD proteins (Fig. 3, red highlighting).

Results of the rate shift analysis for the 19 DEDD proteins. Sites with blue and red highlight correspond to those with slower and faster evolutionary rate, respectively. Sites with entirely blue or red highlight represent amino acid sites with the same evolutionary rate in all families, but with significantly slower or faster rates compared to the average of all sites, respectively.
Furthermore, sequence logo analyses were generated in order to determine the consensus sequence of each of the conserved motifs that were deduced from he alignment of representative deadenylase sequences from both superfamilies (Fig. 4A and B). In this way, a set of structurally-conserved residues were identified on both the DEDD and EEP deadenylases. More specifically, 3 major motifs were identified in DEDD deadenylases and 7 prime motifs in EEP deadenylases (Fig. 4A and B).

Sequence logos of the motifs identified in deadenylase protein sequences. (A) DEDD, numbered according to the human PARN nuclease domain (PDB code 2A1R) and (B) EEP, numbered according to the human CNOT6 nuclease domain. The height of each letter is relative to the frequency of the corresponding residue at that position, and the letters are ordered such as the most frequent is on the top. The invariant catalytic residues that define each superfamily are indicated with dots.
Importantly, apart from the known catalytic residues (Fig. 4A and B), several other residues were found to be evolutionary conserved across species in all various deadenylases. Therefore, these amino acids may serve important functional roles in the action of the deadenylase mechanism. They could also represent potential drug targets.
Footnotes
Author Contributions
Conceived and designed the experiments: AP, DV, NB, SK. Analyzed the data: AP, DV, NB, SK. Wrote the first draft of the manuscript: AP, DV, NB, SK. Contributed to the writing of the manuscript: AP, DV, NB, SK. Agree with manuscript results and conclusions: AP, DV, NB, SK. Jointly developed the structure and arguments for the paper: AP, DV, NB, SK. Made critical revisions and approved final version: AP, DV, NB, SK. All authors reviewed and approved of the final manuscript.
Funding
Author(s) disclose no funding sources.
Competing Interests
NB is supported by the Postgraduate Programs of the Department of Biochemistry and Biotechnology, University of Thessaly, and the Hellenic Thoracic Society. Other authors disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.