
Abstract
It has been known for some time that many proteins are marginally stable. This has inspired several explanations. Having noted that the functionality of many enzymes is correlated with subunit motion, flexibility, or general disorder, some have suggested that marginally stable proteins should have an evolutionary advantage over proteins of differing stability. Others have suggested that stability and functionality are contradictory qualities, and that selection for both criteria results in marginally stable proteins, optimised to satisfy the competing design pressures. While these explanations are plausible, recent research simulating the evolution of model proteins has shown that selection for stability, ignoring any aspects of functionality, can result in marginally stable proteins because of the underlying makeup of protein sequence-space. We extend this research by simulating the evolution of proteins, using a computational protein model that equates functionality with binding and catalysis. In the model, marginal stability is not required for ligand-binding functionality and we observe no competing design pressures. The resulting proteins are marginally stable, again demonstrating that neutral evolution is sufficient for explaining marginal stability in observed proteins.
Introduction
It has been repeatedly observed that a high proportion of globular proteins are marginally stable under physiological conditions, with a ΔGfolding of about −5 to −10 kcal/mol. (Brandts 1967; Privalov and Khechinashvili 1974; Savage et al. 1993; Ruvinov et al. 1997; Vogl et al. 1997; Giver et al. 1998). This is in spite of several factors that suggest that stable proteins might have advantages over marginally stable proteins. For instance, flexible proteins may be less resistant to proteolysis (Fontana, Polverino de Laureto, and De Filippis 1993; Hubbard, Eisenmenger, and Thornton 1994; Fontana et al. 1997; Hubbard, Beynon, and Thornton 1998), denaturation (Wagner and Wuthrich 1979), detrimental conformational change (Carrell and Lomas 1997; Dobson 2001; Bucciantini et al. 2002), aggregation (Lomas and Carrell 2002), and loss of active-site integrity. In addition, binding between less stable and more flexible proteins and their corresponding ligands requires strong binding interactions. More stable proteins do not need such strong binding energies because they lose less entropy upon binding (Schulz 1979). These consequences of higher stability might be expected to increase the evolutionary success of organisms containing stabilised forms of these proteins, suggesting that highly stable proteins should be more common.
The fact that most proteins are not highly stable suggests that other factors are involved, and several hypotheses have been developed to explain this discrepancy. Most of these hypotheses centre on various reasons why marginally stable proteins would have a selective advantage over more stable proteins. For instance, it has been suggested that proteins have evolved towards marginal stability in order to function, suggesting that there is a narrow range of stability consistent with functionality (Rasmussen et al. 1992; Tsou 1998; Zavodszky et al. 1998). There are several reasons why functionality might be limited to proteins of marginal stability. Marginally stable proteins might be more flexible (Wagner and Wuthrich 1979; Tang and Dill 1998), increasing functionality by enabling the formation of strong binding interactions with specific ligands or by providing flexibility needed for conformational change (Lipscomb 1970; Artymiuk et al. 1979; Frauenfelder, Petsko, and Tsernoglou 1979; Wrba et al. 1990; Varley and Pain 1991; Daniel, Dines, and Petach 1996; Zavodszky et al. 1998; Daniel and Cowan 2000). It has also been suggested that marginal stability may be advantageous because ligand binding with marginally stable proteins is comparatively difficult. Binding affinities and specificities of less stable proteins might be more readily adjusted by mutation, phosphorylation or other processes, or selectivity might be enhanced by allowing binding only in the presence of specific interactions (Dunker et al. 1998; Wright and Dyson 1999; Dunker and Obradovic 2001; Dunker and others 2001; Namba 2001). In addition, the physiological importance of marginal stability might involve considerations other than maximizing functionality or selectivity. For instance, unstable proteins might provide more rapid turnover than stable proteins.
A second class of explanations involves the hypothesis that marginal stability is the result of a trade-off between functionality and stability, that the constraints on the amino acids imposed by functionality reduce the opportunities to produce a highly-stable protein resulting in a negative correlation between functionality and stability. This could result if functionality required specific amino acids at functionally-important locations that were incompatible with high stability, so that large numbers of possible sequences, including those with high stability, would be excluded from the evolutionary dynamics. It has been observed, for instance, that mutations increasing protein stability and activity are much more rare than mutations increasing either separately (Alber and Wozniak 1985; Bryan et al. 1986; Liao, McKenzie, and Hageman 1986; Shoichet et al. 1995), although the presence of mutations that increase both (Giver et al. 1998) indicates that functionality and stability are not mutually exclusive. If this trade-off holds for all protein sequences, marginal stability is prevalent because it provides the best balance between the sequences that result in stability and functionality.
These explanations generally arise from an ‘adaptationist’ paradigm, which is to say that the observation of marginal stability in proteins requires an explanation of how this contributes to the reproductive fitness of the organism, either as a direct adaptation or as optimization given constraints. Random events and processes, however, are important factors in the dynamics of evolution and can influence the characters that result (Sueoka 1962; Kimura 1968; King and Jukes 1969). Gould and Lewontin have emphasised the importance of examining possible alternatives to adaptationist selection (Gould and Lewontin 1979). Specifically, they stress that the present usefulness of a character may belie its origins, so that one should avoid ascribing characters to adaptation simply due to their present use. Random events may have led to the existence of the character, which was used to advantage only later. Before adaptation can be judged the cause of the emergence of a character, other explanations must be ruled out.
Consistent with these ideas, a third explanation of the observed marginal stability in proteins involves the concept of regions of sequence space and what has been termed ‘designability’ (Govindarajan and Goldstein 1995; Govindarajan and Goldstein 1996; Li et al. 1996; Buchler and Goldstein 1998; Shakhnovich 1998; England, Shakhnovich, and Shakhnovich 2003). The idea is that the ‘sequence entropy’ or volume of sequence space corresponding to any property influences whether this property is likely to result from evolutionary dynamics. This idea has been applied to the distribution of different structures (Finkelstein and Ptitsyn 1987; Govindarajan and Goldstein 1995; Govindarajan and Goldstein 1996; Li et al. 1996), the question of whether proteins fulfill the thermodynamic hypothesis (Govindarajan and Goldstein 1998), the stability of proteins, (Taverna and Goldstein 2002), ligand binding properties (Blackburne and Hirst 2001; Williams, Pollock, and Goldstein 2001; Blackburne and Hirst 2003; Bloom et al. 2004), and the ability of proteins to explore a range of different structures (Govindarajan and Goldstein 1997a; Govindarajan and Goldstein 1997b; Bornberg-Bauer and Chan 1999; Taverna and Goldstein 2000; Deeds, Dokholyan, and Shakhnovich 2003; Tiana et al. 2004; Shakhnovich et al. 2005). In previous work exploring the evolution of lattice proteins with a fitness function dependent only on stability, it was found that sequence entropy was a sufficient explanation for the observation of marginal stability in proteins, and that this effect would favor mechanisms of function consistent with marginal stability (Taverna and Goldstein 2002). In further work, we developed a simple model with fitness represented by the ability of the protein to bind a ligand, observing similar results (Williams, Pollock, and Goldstein 2001).
To examine these hypotheses in the context of protein functionality and to extend the previous work, we simulate the evolution of proteins using a lattice-protein model of protein-folding and ligand-binding that allows the study of protein stability and function. In these simulations, the fitness function is based on diffusion-limited reaction kinetics. The model is designed so that fitness increases with binding strength, which tends to increase with stability, meaning that marginally stable proteins have no evolutionary advantage over proteins of greater stability. We also observe no design constraints or trade-offs between stability and functionality among these evolved proteins. Our evolutionary simulations, however, lead to marginally stable proteins. This indicates again, with a more realistic simulation, that random evolution is sufficient to generate marginal stability. This does not prove that marginal stability is not an adaptation, but rather demonstrates that marginal stability could result in the absence of any adaptive role, that its presence does not indicate that it plays such an adaptive role, and therefore that marginal stability is not a phenomenon that needs an explanation based on evolutionary advantage.
Methods
Protein Model
Protein models should accurately represent important and relevant aspects of real proteins yet be simple enough for rapid computational evaluation. Our model must be relatively simple indeed, as evolution simulations involve the examination of many protein sequences over a large number of generations. To examine the previously described hypotheses, the model must map protein sequence to stability in a compact state as well as the propensity for binding and acting upon a specified ligand.
The details of this model have been more thoroughly described elsewhere (Williams, Pollock, and Goldstein 2001). Each model protein consists of a chain of 16 amino acids on a 2-dimensional square lattice. While the 2-dimensional model is problematic for dynamics simulations (Shakhnovich 1997), for thermodynamic analyses involving sums over states it is more accurate at representing the appropriate number of buried vs. exposed residues. Intra-protein contacts are defined as non-sequentially-adjacent residues one lattice-unit apart in distance. Compact structures have nine contacts (the maximum number possible for a 16-residue protein) and fit in a square with four residues per side. All 802,075 possible structures are considered, of which only 69 are compact.
The free energy G(k) of a sequence {A1, A2…A16} in conformation k is given by

We use Boltzmann statistics to determine P(k), the thermodynamic probability of folding into conformation k, assuming all conformations are in equilibrium:
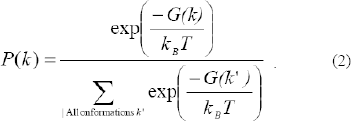

We model protein-ligand binding as a four-residue peptide contacting any of the four sides of a compact protein, such that maximal contact between the ligand and the face of the protein is made, as illustrated in Figure 1. The ligand may face either of two directions on any of the four sides of a conformation, so there are 69 × 4 × 2 = 552 possible binding sites on a protein sequence. The free energy of a complex where the protein is in compact conformation k and the ligand is bound to site l is
An example of a model protein in a compact conformation bound to a ligand, shown in grey. Covalent bonds are shown as solid lines, contact interactions as thick (intramolecular) and thin (intermolecular) stripes.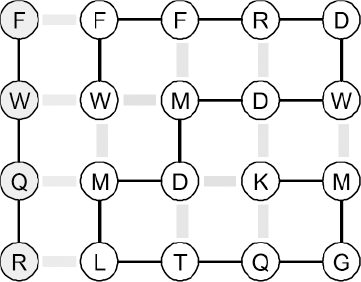


Under conditions when very little of the protein is bound to ligand, we can ignore the second term in the denominator and calculate the relative probability of the protein binding the ligand for a fixed concentration by multiplying P0(Bound,) by exp(–ΔSlig/k
B
), yielding

Evolution Model
We model a population of random proteins evolving through mutation and replication. Starting with an initial population of 1000 protein sequences, we allow a fixed rate of mutations, modeled as a Poisson distribution with a mean of 20 mutations per generation. Sequences are then replicated according to their fitness, as described below. The population size is maintained at a constant level of 1000 proteins throughout the experiment.
Measure of Fitness
Many factors affect the evolutionary success of a protein, ranging from the intrinsic properties of the protein to external and indirectly related circumstances. For this paper, we are concerned only with the effects of selection for protein functionality. To study these effects, we construct a fitness function based on the rate of catalysis of bound ligands.
Assuming that product-formation is beneficial, we consider fitness directly proportional to the rate of catalysis. We model this rate with Michaelis—Menten kinetics, corresponding to reactions of the following type,

We are interested in investigating the ease with which marginal stability could result in evolving proteins in the absence of any specific advantage to marginal stability. For this reason, we assume that k2 is independent of binding strength. In this case, the rate of catalysis, and thus the fitness used in the evolution simulations, is given by

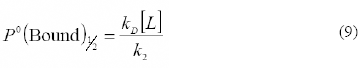
Fitness increases monotonically with increasing
In our evolution runs, proteins are selected for their ability to bind a specific ligand. The sixteen four-residue permutations of glutamate and lysine can form the strongest binding interactions of any ligand, while the optimal binding interaction of a polyalanine ligand is the weakest possible. We performed evolution runs using the ligands AAAA, EEEE, and EKEK, to examine the influence ligand-choice has on evolution. Real proteins have a wide range of k2 values, and act on ligands of varying concentration, diffusion rates, and ΔSlig. To account for this variety, we performed simulations with a range of values for
In addition to performing evolution experiments, we also optimise proteins for maximum fitness. Beginning with a random sequence and a specified ligand, we perform hill-climbing walks on the fitness landscape. The steps made on this landscape are random point mutations of the protein sequence; a mutation is accepted if it results in an increase in the fitness, that is to say, an increase in
Results
The results of a typical evolution run, showing population-weighted averages of P(Compact) and Extended typical evolution run with fitness based on ability to bind and catalyze EEEE, showing the effects of changing 
Figure 3 illustrates the physical properties of the final generations of the evolution experiments for two different ligands, AAAA (3a-3b) and EEEE (3c-3d), compared with the proteins that result from optimization through hill-climbing. Results for different values of The properties of evolved and optimised proteins, colour-coded by the value of 
There are several possible explanations for the relationship between overall protein stability and probability of binding a ligand. Thermodynamically, we would expect that a protein with higher stability would, on average, bind more strongly than unstable proteins, due to the entropy penalty when a less-stable protein binds a ligand. Alternatively, the ‘optimization given constraints’ model suggests that there might be a negative correlation between protein stability and binding, as the residues that optimized stability might not be the same as the residues that optimised binding interactions. The positive correlation between binding probability and protein stability shown in Figures 3b and 3d, demonstrates that the thermodynamic effect dominates any possible trade-offs between stability and binding.
Further evidence of the lack of trade-off between ligand binding and protein stability is provided by considering the proteins that have been optimised for binding by the hillclimbing algorithm. We observe no correlation between the stability of the proteins that result and the strength of their binding interactions with the peptide ligand, as would be expected if strong binding interactions were incompatible with high protein stability.
In general, the properties of evolved proteins fall within a range of values, but the ligand and the
These differences are due to the nature of the ligand. A mutation can increase
Discussion
Figure 3 shows that most proteins resulting from our evolutionary experiments are not highly stable, consistent with observations about real proteins. In fact, most of the resulting proteins tend to have low or marginal stability levels, depending on ligand and value of
As described in the Introduction, there are two different possible reasons generally given for the marginal stability of real proteins - either a specific selective advantage of marginal stability, or a co-optimization of the conflicting qualities of stability and other aspects of functionality. In our model, we have eliminated the first possibility by construction, in that proteins that bind more strongly have a higher fitness. We also observe a positive correlation between stability and fitness, so the theory of conflicting design pressures does not apply in our model. With both of these possible explanations inappropriate for these simulations, we still observe marginally stable proteins.
This provides evidence that sequence entropy, the third possible explanation, is sufficient to explain the observation of marginal stability in biological proteins. We are not ruling out the other proposed explanations, but we can explain the observed properties of evolved proteins without them. The existence of marginal stability in real proteins implies neither an evolutionary advantage to marginal stability nor a trade-off between stability and binding strength. The most parsimonious explanation for marginal stability does not include either of these two mechanisms.
The protein models used in this study are smaller than realistic proteins, and thus might better represent the area surrounding an active site more than an entire protein. Of the three explanations used to explain marginal stability - evolutionary advantage of marginal stability, negative correlation between stability and fitness due to design constraints, and sequence entropy - we would expect the first explanation to be independent of protein size, the second explanation to be especially appropriate around the active site, while the third explanation would involve the entire protein, as the protein generally is required to be folded in order to bind and catalyse a ligand. The fact that we do not observe evidence for the second explanation while the third explanation seems to be adequate in these smaller models suggests that it should also be adequate when a more realistically-sized protein is considered.
The effect of the underlying nature of protein sequence space may have been more important in early protein evolution. Most proteins with random amino-acid sequences are highly unstable (in our model, and likely in the real world), so the first existing proteins would likely have been unstable as well. Some degree of stability was likely beneficial, so mutations that lead to increased stability were accepted. At a certain point, the fitness gains of higher stability were counter-acted by the effect of sequence entropy, and thus protein stability did not increase further.
The resulting stability depends upon the ligand, as well as
Footnotes
Acknowledgements
Thanks to Darin Taverna for helpful discussions and Todd Raeker for computational assistance. Financial support was provided by the Medical Research Council and by NIH grant ROI LM005770.
Appendix: Derivation of the fitness function
To derive the fitness function, we start with equation 8. In the Michaelis-Menten model, the rate of change of [PL] is assumed to be zero, so the steady state concentration of PL is
The total concentration of protein, [P]
T
, is equal to [P]+[PL]. Solving for [P] and [PL] in terms of [P]
T
yields
The rate of production of the final product is
We can relate this to terms calculated from our protein model by expressing P0(Bound), the relative thermodynamic probability that the protein binds a ligand, in terms of protein and ligand concentrations. P0(Bound) = [PL]/[P]T, under conditions when there is no forward reaction, or k2=0. Under these conditions, equation 13 becomes,
Solving for kuni yields
Substituting this expression into equation 14 yields
We assume that k
D
, [P]
T
, and [L] remain relatively constant, and as fitness is relative and thus not changed by a multiplicative factor, the final fitness function is then:
where P0(Bound)1/2 = k D [L]/k2 is the value of P0(Bound) where the fitness is half the maximum fitness.