
Abstract
Microbiomes are ubiquitous and are found in the ocean, the soil, and in/on other living organisms. Changes in the microbiome can impact the
Introduction
Communities of microbes are found in diverse environmental niches, such as the ocean, soil, and inside host organisms, including all animals, plants, and lower eukaryotes.
1
These communities show characteristics, such as complexity, diversity, interaction, cooperation, dynamism, generosity, danger, and competition.
2
In such communities, microbes may compete for nutrients,
3
share functional genes through horizontal gene transfer,
4
produce toxins that can kill other microbes,
5
produce various metabolites and signaling molecules for sharing and communication,
6
and combine forces to fight common enemies, such as the host immune system.
7
In short, the importance of the microbial community stems from the fact that they are critical to the
Traditionally, a microbiome has been defined as a microbial community occupying a reasonably well-defined habitat. 10 One of the most common approaches to studying a microbiome is analyzing its constituent microbial genomes through metagenomics. More recently, this definition has evolved to include not only the microbes and their genomes but also the aggregate of environmental and host factors. The inclusion of the host environment as part of the microbiome significantly expands its implications, with the interactions between the host and its associated microbial community now relevant to understanding the dynamics of the microbiome. For evolutionary and functional studies of the microbiome, modifications in the host environment (eg, a diet shift in the host organism or a compositional change in the environmental matrix under study) now become critical and must be taken into consideration. Coevolution processes can then be identified, providing valuable information to understand the relationship of the microbial community with its host. This apparent conceptual shift is accompanied by the recognition that, in order to achieve a more comprehensive study of microbiomes, metagenomics must be combined with other omic approaches. Many relevant omic approaches have been proposed for microbiome studies. In this article, we discuss metatranscriptomics and metabolomics, which are rapidly becoming critical to microbiome studies.
Metagenomics is the study of the genomes in a microbial community and constitutes the first step to studying the microbiome. As seen in the “Metagenomics” section, metagenomics comes in different flavors. However, its main purpose is to infer the taxonomic profile of a microbial community. Although whole-metagenome sequencing (WMS) provides a partial glimpse into the functional profile of a microbial community, it is better inferred using metatranscriptomics, which involves sequencing the complete (meta)transcriptome of the microbial community. Metatranscriptomics informs us of the genes that are expressed by the community as a whole. With the use of functional annotations of expressed genes, it is possible to infer the functional profile of a community under specific conditions, which are usually dependent on the status of the host. While metagenomics helps address the question “what is the composition of a microbial community under different conditions?”, and metatrascriptomics helps answer the question “what genes are collectively expressed under different conditions?”, the question considered by metabolomics is “what byproducts are produced under different conditions?”. The metabolites released by the microbial community are largely responsible for the health of the environmental niche that they inhabit.
Regardless of whether microbiome studies are biomedical or environmental in their focus, it is clear that the different omic approaches provide invaluable information. However, the best results are obtained by performing
As suggested by a variety of different analyses,13–16 we believe that network-based approaches can lead to a sophisticated in-depth analysis of microbiomes, particularly when applied to integrative studies, and consequently lead to critical insights into the world of microbiomes.
Major microbiome initiatives
Human microbiome studies
The National Institute of Health has funded a major initiative that aims to generate resources for a comprehensive characterization of the human microbiome to understand its impact on human health and disease. The first phase, known as the Human Microbiome Project (HMP), 17 focuses on the study of microbial communities that inhabit the human body of healthy individuals,18,19 with particular emphasis on nasal, oral, skin, gastrointestinal, and urogenital areas.17,18,20–23 It is known that the amount of microbial cells present in the human body is notably larger than the amount of human cells. These bacterial communities play critical roles, such as assisting in the digestion of food, synthesizing necessary vitamins, and aiding the immune system in defending our body from pathogenic invaders. 24 Human microbiome studies have revealed strong correlations between changes in microbial community profiles and diseases.22,25–27 These studies have also shown that the structure of the microbial community is significantly different in five areas of the human body (gut, mouth, airways, urogenital, and skin), and that this seems to be independent of gender, age, and ethnicity.18,19 All the data and protocols associated with this project are available at the HMP Data Analysis and Coordination Center (DACC). 28
The Integrative HMP (iHMP) 27 is the second phase of this initiative, going a step further by gathering multiple omic data from both the microbiome and the host. This is part of a longitudinal study with a broader objective of understanding host-microbiome interactions using integrative analyses. Another related initiative focused on the human microbiome is the Metagenomics of the Human Intestinal Tract (MetaHIT) project. 29 This project was funded by the European Seventh Framework Programme until 2012. Its goal was to understand the link between the human intestinal microbiota and human health/disease. For this purpose, they focused on two disorders of increasing incidence in Europe: obesity and inflammatory bowel disease. Similarly, the Human Food Project and the American Gut Project 30 focus on the gut microbiome with the aim of determining how to acquire a healthy microbiome through food.
Environmental microbiome studies
The Earth Microbiome Project (EMP) is a remarkable effort started in 2010 to characterize the diversity, distribution, and structure of microbial ecosystems across the planet and has already gathered over 30,000 samples. 31 Their focus is on diverse ecosystems, including not only the ones within the bodies of humans, animals, and plants but also terrestrial, marine, freshwater, sediment, air, and constructed environments, as well as every intersection of these ecosystems.
J. Craig Venter Institute's (JCVI) Global Oceanic Sampling (GOS) expeditions and the European Tara Oceans initiatives32–36 have focused on understanding and cataloging the marine microbiome diversity across the planet. JCVI's vessel,
Metagenomics
Metagenomics allows us to investigate the composition of a microbial community.
Recently, some authors have argued in favor of a terminological distinction between metagenomics (used to describe a broad comprehensive genomic approach to microbiome profiling) and metataxonomics (which uses amplicons from a targeted marker gene in order to make taxonomic inferences).
37
One popular marker gene used in metataxonomic studies is
Tools and techniques
A variety of tools and analysis pipelines have been developed to analyze metagenomic data.
50
problem solving environments (PSEs
51
) provide user-friendly workbenches to develop flexible scientific analysis pipelines using a menu of available tools. Such workbenches incorporate different ranges of generality. For instance, Galaxy
52
maximizes generality by providing a framework for genomic analysis while allowing the user to supply tools and file formats for various stages in a pipeline. Galaxy can execute jobs remotely, allows for
As shown in Figure 1, metagenomic analysis pipelines can be divided into three main steps: (1) preprocessing the reads, (2) processing the reads, and (3) downstream analyses.

Generic microbiome analysis pipeline.
Preprocessing and processing the reads
The procedures followed in preprocessing and processing of the reads (steps 1 and 2) have become fairly standardized. Hence, we describe them briefly and focus mostly on downstream analysis (“Downstream analyses of metagenomic data” section).
Preprocessing mainly involves removing adapters from reads, filtering reads by quality and length, removing contaminants, identifying and removing any chimeric sequences that may have been generated during polymerase chain reaction (PCR) amplification, and preparing data for subsequent analysis. A survey of some of the popular tools and techniques currently available for this step can be found in Kim et al. 50
After preprocessing of the reads, the next step is to classify each read based on the taxa with the highest probability of being the origin of that read. This step often uses a reference database of relevant microbial genomes and produces a microbial profile usually represented as an abundance matrix with microbial taxa as rows, samples as columns, and values representing the abundance of a taxon in the sample.
In the case of metataxonomics, reads are frequently grouped (or clustered) prior to assigning a label. Unlike WMS, which produces a lower coverage and may identify thousands of strains per sample, targeted approaches have reads that come from relatively small regions of the genome, making this extra clustering step valuable in lowering errors in the classification. Groups of reads that result from the clustering process displaying similarity in sequence and/or composition are inferred to have a common origin and referred to as operational taxomonic units (OTUs).
The classification and labeling performed on the reads can be either taxonomy dependent or taxonomy independent. Taxonomy-dependent methods use a database of reference genomes, which has some bias toward data with pathogenic or commercial applications. Methods in this category can be further classified as alignment-based, composition-based, or hybrid. Alignment-based methods usually give the highest accuracy but are limited by the reference database and by the alignment parameters used and are generally computation and memory intensive. Composition-based methods store only compact models instead of the whole genome, requiring fewer computational resources. These methods use features extracted from the genomes (eg, GC percentage and codon or oligonucleotide usage patterns) to build models but have not yet achieved the accuracy of alignment- based approaches. Hybrid approaches offer a compromise between the two. Taxonomy-independent methods, on the other hand, do not require a priori knowledge. Instead, they segregate reads based on properties, such as distance, k-mers, abundance levels, and frequencies. These methods are typically used if the samples are more likely to have microbes that are not documented in the databases. Chen et al. 56 and Mande et al. 57 reported an extensive review of popular tools and techniques used for processing 16S reads and for processing WMS reads, respectively.
Accurate classification and labeling are challenging because (a) sequencing technologies produce short reads, (b) for economic reasons the datasets often have low coverage of the genomes in the microbiome, (c) some sequencing technologies have a high percentage of sequencing errors, and (d) the reference genome databases used are not comprehensive, often failing to provide an accurate taxonomic context because of lateral gene transfers between microbial taxa.
Downstream analyses of metagenomic data
Once the reads have been assigned labels or classified as best as possible, downstream analyses attempt to extract useful knowledge from the data. Typical questions addressed in this step include “how diverse are the microbial taxa in the sample?”, “what is the functional profile of the genes present and/or expressed in the microbial community?”, “what microbial taxa are differentially abundant in the samples?”, “what phylogenetic groups, functional and metabolic pathways, orthologous groups of genes, and gene ontology terms are particularly enriched or depleted in the samples?”, and “what microbial groups tend to co-occur or co-avoid in the samples of interest?”. We now review several current tools and techniques for performing downstream analysis.
Richness and diversity are measures that have traditionally been used to characterize a metagenomic sample.58,59 Richness is a simple count of taxa present in a sample. Diversity refers to a collection of indices and measures (eg, Shannon, Chao, Simpson, and Berger-Parker) that quantify the evenness of the distribution of the abundances of the taxa, 59 often incorporating distance measures or similarity indices (eg, Jaccard, Sorenson, and Bray-Curtis). Richness and diversity offer measures of complexity of the community but disclose little about interactions within the community, which requires more complex downstream analyses.
Visualizing taxonomic profiles is a task that has been addressed by several initiatives. Krona, 60 for example, is a simple and intuitive web-based tool to visualize the taxonomic profile as a pie chart with an embedded hierarchy. In contrast, the Visualization and Analysis of Microbial Population Structure (VAMPS) tool 61 can measure and visualize statistically significant similarities and differences between multiple taxonomic profiles of complex microbial communities.
Integrating additional information in metagenomic analyses is extremely valuable in order to provide improved perspectives of the microbial profiles. Based on this premise, a number of approaches have sought the use of phylogenetic information to enhance the labeling and classification of reads, as is the case with Amphora2, 62 which performs phylogenetic inference using phylum-specific marker databases. This type of inference can be done algorithmically as well, through edge principal component analysis (PCA) and squash clustering. 63 Phymm64,65 is a software package that classifies sequence fragments into phylogenetic groups using interpolated Markov models. Finally, PPlacer 66 performs phylogenetic placement using a fixed reference tree and maximum-likelihood inference with distance calculations to indicate uncertainty and can be executed in parallel.
A more significant improvement is possible with the help of functional annotations of the genes to which the reads are mapped.67,68 Although many analytical metagenomic approaches focus on the composition or structure of the samples, functional profiling is also essential, as it provides insight into the underlying biological processes. Other useful resources for annotation include gene ontology (GO),69,70 Kyoto Encyclopedia of Genes and Genomes (KEGG),71,72 and Clusters of Orthologous Groups (COG).73,74 As a part of the HMP initiative to analyze WMS data, a methodology called HUMAnN 75 was developed for inferring the functional and metabolic potential of a microbial community.
Alternatively, other existing tools, such as IMG/M, 76 CAMERA, 77 METAREP, 78 MEGAN, 79 and CoMet, 80 can also be used to obtain functional profiles of microbiomes. IMG/M, METAREP, and CoMet provide a web-based user interface, while CAMERA aims to offer a state-of-the-art computational structure for high-performance network access and grid computing as a part of a distributed architecture. In contrast, MEGAN is a standalone computer program. METAREP and CoMet annotate the data with GO and KEGG, whereas MEGAN uses the NCBI taxonomy to summarize and order the results obtained after performing BLAST. METAREP also offers the option to annotate the data with taxonomic information, and IMG/M uses BLAST to infer phylogenetic information from the sample. However, IMG/M is more oriented toward protein-related information by annotating the results with resources, such as COG, Pfam, TIGRFAMs, ENZYME, and KEGG. IMG/M was developed by the Joint Genome Institute and contains data from the HMP and the Genome Encyclopedia of Bacterial and Archaea Genomes. CAMERA has been designed for environmental and ecological purposes with the aim of providing new ways of visualizing and interacting with data and was applied to data from GOS. METAREP, on the other hand, was developed at JCVI. It performs statistical tests and muti-dimensional scaling (MDS) and can also produce graphical summaries, heatmaps and hierarchical clustering plots. MEGAN uses the lowest common ancestor algorithm to label the reads and has been applied to datasets, such as the Saragaso Sea dataset, and data from mammoth bone. Finally, CoMet combines open reading frame finding and assignment of protein sequences to Pfam domain families with comparative statistical analysis, providing the user with comprehensive tabular data files and visualizations in the form of hierarchical clustering and MDS. It was applied to 454 data.
Obtaining the functional profile is typically not possible with targeted approaches, since it provides no direct evidence of the functional capabilities of the microbial community. However, the tool Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) shows how to infer a functional profile of a microbial community directly from taxonomic profiles of marker genes, such as the
Discussion
In summary, metataxonomics helps us to compute the taxonomic profile of a microbial community, while metagenomics helps us to compute the functional profile by focusing on the gene content and using the available functional annotations of the corresponding proteins. While metagenomics is powerful, solely using it to study a microbiome is limited in value. Many experts have confirmed that the percentage of documented bacteria is very low compared to the estimate of bacterial species on our planet.
82
This may be due partially to the impossibility of culturing complex environments or replicating in the laboratory the real conditions in which the microbiome exists. Either way, the reference databases used to classify and label bacteria are limited to what has been cataloged. Current methods typically either discard reads from undocumented microbes or label them based on the
Metatranscriptomics
By focusing on what genes are expressed by the entire microbial community, metatranscriptomics sheds light on the
When processing reads, a typical metatranscriptomics analysis pipeline will either (1) map reads to a reference genome or (2) perform de novo assembly of the reads into transcript contigs and supercontigs. The first strategy, in a manner similar to the alignment-based methods in WMS, maps reads to reference databases, thus gathering information to infer the relative expression of individual genes. The second strategy infers the same but with assembled sequences. The first strategy is limited by the information in the database of reference genomes. The second strategy is limited by the ability of software programs to assemble contigs and supercontigs correctly from short reads data.
Tools and techniques
The application of metatranscriptomics to the study of the microbiome is far less common relative to other omics reviewed in this article. Most analysis pipelines described in the literature were built ad hoc. The majority of these methods follow the aforementioned first strategy based on read mapping.88–92 In this case, metatranscriptomic reads are generally mapped to specialized databases (usually downloaded from the NCBI) using alignment tools, such as Bowtie2, BWA, and BLAST. The results are then annotated using resources, such as GO, KEGG, COG, and Swiss-Prot. Finally, different types of downstream analysis are carried out depending on the goal of the study (eg, PCA-based phylogenetic analysis or enrichment analysis). The latest metatranscriptomics techniques include stable isotope probing (SIP), which has been used to retrieve specific targeted transcriptomes of aerobic microbes in lake sediment. 93 This not only helps to target specific organisms but also contributes significantly to metabolomics studies.
The second strategy requires assembling metatranscriptomic reads into longer fragments called contigs. For this purpose, numerous software packages are available. Celaj et al.
94
compared de novo sequence assemblers to reference-based
Discussion
Although current metatranscriptomic techniques are promising, there are still several obstacles that limit their large-scale application. First, much of the harvested RNA comes from ribosomal RNA, and its dominating abundance can dramatically reduce the coverage of mRNA, which is the main focus of transcriptomic studies. Some efforts have been made to effectively remove rRNA.
107
Second, mRNA is notoriously unstable, compromising the integrity of the sample before sequencing. Third, differentiating between host and microbial RNA can be challenging, although commercial enrichment kits are available. This may also be done
WMS approaches provide information on the taxonomic profile of a microbial community as well as its potential functional profile; in contrast, whole metatranscriptome sequencing describes the active functional profile. This would help in studying the dynamics of functional profiles with varying conditions. We now discuss metabolomics, which studies the consequences of the shifts in the collective gene expression of the microbial community that modifies the very medium where the microbial community must feed, grow, reproduce, and cooperate or compete to survive.
Metabolomics
Metabolomics is the comprehensive analysis by which all metabolites of a sample (small molecules released by the organism into the immediate environment) are identified and quantified. 109 The metabolome is considered the most direct indicator of the health of an environment or of the alterations in homeostases (ie, dysbiosis). 110 Variation in the production of signature metabolites are related to changes in activity of metabolic routes, and therefore, metabolomics represents an applicable approach to pathway analysis. 111 Additionally, the application of metabolomics for drug discovery and pharmacogenomics represents a promising avenue for personalized medicine. 112
The metabolomic profile associated with the microbiome may show a strong dependence on environmental factors (eg, diet, exposure to xenobiotics, and environmental stressors), providing valuable information not just about the characteristics of the microbiome but also about the interactions of the microbial community with the host environment.113–115 Thus, metabolomics aims to improve our understanding of the role of the microbiome in the transformation of nutrients and pollutants as well as other abiotic factors that may affect the homeostasis of the host environment. Microbial communities exert a strong influence on critical biogeochemical cycles, and the study of their metabolome can help to develop predictive biomarkers for environmental stressors. 116 The microbiome is regarded as a biological reactor that, based on its genetic pool, can transform resources and hazardous elements into products that are either beneficial or detrimental to the health of its environment. A good example is bioremediation and its application to reduce the consequences of pollution. 117
Most interestingly, the metabolome can illustrate signaling processes involved during communication between bacteria, such as quorum sensing, which relates gene expression responses to changes in cell population density.118–123 A deeper understanding of the communication mechanisms within microbial communities could possibly revolutionize the current strategies in areas such as infections disease control, and optimize agricultural exploitation in environmental conservation. Thus, metabolomics complements the information provided by the other omics (mentioned earlier) by describing not just biological systems themselves, but how they interact internally and externally.
Generating metabolomics data differs significantly from generating metagenomics and metatranscriptomics data, which rely heavily on sequencing. Identifying and quantifying metabolites is typically carried out using a combination of chromatography techniques (ie, liquid chromatography, LC, and gas chromatography, GC) and detection methods, such as mass spectrometry (MS) and nuclear magnetic resonance (NMR). For a more detailed review of these technologies and their many variants, we refer the reader to a recent review by Aldridge and Rhee. 124 These technologies produce spectra consisting of patterns of peaks that allow both the identification and quantification of metabolites. These patterns (either predicted or experimentally obtained) are stored in spectral databases, allowing automated analysis and generation of metabolomic profiles. With these technological resources, metabolomics fulfills the requirements of a high-throughput analytical method, and thus data analysis represents a critical step in knowledge generation. As a result, we have seen a rise in software development, large data repositories, and initiatives for standardization. This in turn paves the road for data integration.
Tools and techniques
The analysis pipeline for spectral metabolomic data involves three steps: (1) preprocessing, (2) statistical analysis, and (3) machine learning techniques for pattern recognition. 125 In the first step, denoising and peak-picking improve the quality of the data to be processed. Once the peak pattern has been established, a comparison against spectral databases identifies the metabolites in the sample and the area below the peaks their respective quantities. To automate this process, spectral databases are maintained and curated by specialized international consortia that emphasize standardization. These include the following: the Human Metabolome Database, a cross-referenced database about the small metabolites found in the human body126–128; the BioMagResBank, which works as a central repository for experimental NMR data including both small metabolites and macromolecules 129 ; the Madison-Qingdao Metabolomics Consortium Database, 130 which includes both NMR and MS data thoroughly annotated collected from other databases and literature; MassBank, 131 which merges spectral data from different collision-induced dissociation conditions to improve the precision in the identification of compounds; the Golm Metabolome Database, 132 which stores spectral data with retention indexes, useful for automated identification of compounds analyzed with GC-MS; and the METLIN Metabolite Database, 133 which contains curated spectral information of biological metabolites without information of the environmental context from which the samples where obtained. Each of them differs slightly in functionality but pursues similar goals, serving as repositories of spectral data and offering links to their biological interpretation.
Discussion
By cataloging all metabolites present in a sample, metabolomics offers a powerful way to relate the metabolites to the cellular processes of which they are the byproducts. The combination of metabolomic and pathways information can lead to new hypotheses. One important challenge of this approach is difficulty in determining whether a metabolite was generated by the host or by the microbiome. In addition, if conclusions are to be made about which genes, enzymes, or pathways are associated with a specific metabolite, the results obtained from a metabolomic study must be combined with other omic data. This highlights the need for new approaches that deal with integrated omics, as discussed in the “Integrating multiomic data” section.
Integrating Multiomic Data
Standard analyses of individual omic datasets focus on the community structure and functional roles of individual taxa or groups of taxa. The remaining challenge lies in elucidating the large, dynamic, and complex network of interactions between its constituent entities. With the increasing availability of heterogeneous multiomic datasets,
11
the need for integrative analyses has become even more urgent. A reasonable approach (Fig. 2) is to perform separate analysis, adding an extra integrative step within
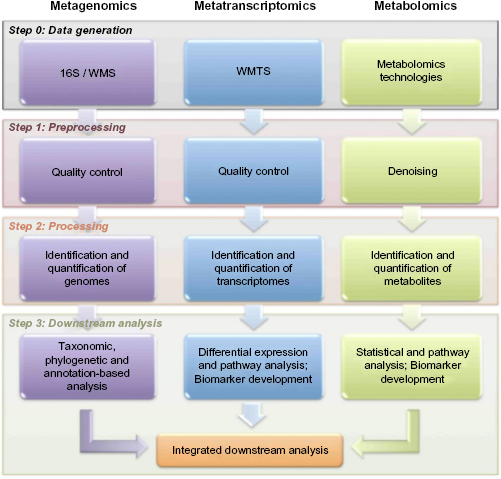
Generic multiomic analysis pipeline.
Integrating multiple omic datasets is a problem that researchers are just beginning to tackle. 12 Bringing together different studies will allow researchers to build and test mathematical models of microbial activity and interaction, enabling a better understanding of the interplay between the environment and the microbial community.134,135 For example, the combination of metagenomics and metatranscriptomics may reveal overexpression or underexpression of particular functions and, in some cases, the activities of specific organisms.90,136–138 The addition of metabolomics could provide insight into the outcome of those changes in gene expression, which may lead to differential expression of specific metabolites that impact the health of the host environment.139–144 Understanding the whole ecosystem opens new avenues and exciting approaches for generating new knowledge. By combining multiple (potentially noisy and heterogeneous) data types, we can build support for specific hypotheses; if independent lines of evidence arrive at the same conclusion, then our confidence in that conclusion will grow.
Tools and techniques
Current studies indicate that integrating metagenomics and metatranscriptomics has the potential of attributing functional changes in gene expression to specific members of the microbial community. Franzosa et al. 145 showed a relationship between genomic abundances and differential regulations of microbial transcripts, discovering up- and downregulated pathways within the human gut microbiome. Shi et al. 146 applied this integrative approach relating the functional and taxonomic profiles of marine environmental samples. Current studies also indicate that integrating the results of metagenomics with metabolomics can provide insight into how members of a microbial community interact with each other and with their environment. 147 For example, Lu et al. 148 observed a simultaneous effect on both microbiome composition and metabolite production upon introducing arsenic into the mouse gut environment. Zhang et al. 149 performed a similar study with the introduction of disinfection byproducts from drinking water. These studies illustrate that the different omics are interdependent and that an integrated approach can lead to more useful discoveries.
Several current studies suggest that integrating all three omic data – metagenomics, metatranscriptomics, and metabolomics – would provide a complete picture from genes to phenotype.150,151 With the wealth of datasets available but not currently integrated, Abram 152 argues for a system-based approach to multiomics, which would allow predictive modeling. In particular, he points out that studying interrelationships between entities (which he refers to as SIP-omics) would provide some guidance to establishing linkages between various datasets.
Interrelationships also form the basis of the reverse ecology algorithm,
153
which attempts to connect microbial communities with properties of their environment under the assumption that adaptation to the environment is most fundamental to their structure and topology. The set of metabolites that are acquired by an organism from external sources is called the
Conclusion and Future Directions
In this article, we have discussed how three different omic approaches – metagenomics, metatranscriptomics, and metabolomics – provide useful information toward understanding microbiomes. We also discussed how the value of an integrative approach is greater than the sum of its parts.
Biological networks have long been used to model interactions between biological entities, with applications to areas, such as gene regulation, metabolic and signaling pathways, protein-protein networks, and food webs in ecology.156–159 With its proven application to analyzing interrelationships and their critical role in multiomics, we believe biological network analysis will be critical to future multiomic approaches to studying the microbiome. In addition, network analyses offer the possibility of exploring both local (eg, relationship with neighbors) as well as global properties (eg, connectivity) of a community. Dutkowski et al. 160 studied the assignment of ontologies using networks and developed tools, such as Cytoscape, 161 to perform these analyses.
Metagenomic studies have shown that interactions within a microbiome can be naturally modeled using a network representation,14,42,162 with properties closely related to
In order to integrate data from various omic sources, microbiomes can also be modeled as heterogeneous networks (Fig. 3), which provides a visual description of what such a network in the context of the microbiome would look like. A heterogeneous network would allow researchers to generate new interesting hypotheses that involve entities from the different omics described in this article (represented in the figure by nodes with different shapes and colors). For instance, we could potentially have a

Integrated networks for multiomic data.
Footnotes
Author Contributions
Conceived and designed the experiments: VAP, GN. Analyzed the data: VAP, WH, VSU, TC, GN. Wrote the first draft of the manuscript: VAP, WH, VSU, TC. Contributed to the writing of the manuscript: VAP, WH, VSU, TC, GN. Agree with manuscript results and conclusions: VAP, WH, VSU, TC, KM, GN. Jointly developed the structure and arguments for the paper: VAP, GN. Made critical revisions and approved final version: VAP, KM, GN. All authors reviewed and approved of the final manuscript.