
Abstract
Recent research on genome evolution of large DNA viruses has highlighted a number of incredibly dynamic processes that can facilitate rapid adaptation. The genomes of amphibian-like ranaviruses - double-stranded DNA viruses infecting amphibians, reptiles, and fish (family
Introduction
Large DNA viruses have traditionally been viewed as somewhat evolutionarily inactive, exhibiting low substitution rates and cospeciation alongside their hosts. 1 This may have sprung from an overemphasis on the effect of the molecular mechanisms of replication (eg, the specific enzymes utilized and the resultant error rates, which ultimately determine mutation rates) and a lack of consideration of other biological factors that determine substitution rates such as generation time, life cycle, and selection. 2 Indeed, although phylogenetic concordance between hosts and viruses has been observed frequently, in some cases it has been shown to stem from preferential host switching rather than cospeciation. 3 Several lines of evidence from recent studies show that large DNA virus genomes can be highly dynamic. Genome evolution via lateral gene transfer occurs frequently with examples of the acquisition of genetic material from eukaryotes, bacteria, and bacteriophages.4–8 Double-stranded DNA viruses exhibit considerable variation in substitution rates. 2 Viruses such as the variola virus (a poxvirus causing smallpox) appear to have substitution rates on par with RNA viruses, and there is tentative evidence for high rates in other virus groups too.9,10 Recombination between viruses is also now known to be extremely widespread in some large DNA viruses.11,12
Nucleocytoplasmic large DNA viruses form a monophyletic group of seven virus families (
The family
ALRVs with sequenced whole genomes are geographically widespread and have been isolated over a 50-year period. Genomes range from 100 to 130 kb in size and contain approximately 100 genes with low functional annotation rates.18,29 The ALRVs form a distant sister clade to grouper iridovirus-like ranaviruses (GIV-like; pathogens of marine fish) and are frequently divided into three groups:
Methods
Data
Sixteen published ALRV genomes were downloaded from NCBI nucleotide database (full GenBank [.gbk] format and the nucleotide sequence only format [FASTA]) (Table 1). These viruses were isolated in the USA, Europe, China, and Australia over a 50-year period from 1965 to 2013 (Table 1). The same files were also downloaded for the GIV-like ranaviruses for use as outgroups and in some comparative analyses (Table 1).
Isolation history and source of data for

For some isolates, the year of collection is provided in the GenBank record or accompanying publication. For others, the year of collection is estimated from other information in the accompanying publication, and this is indicated in the table by appending (approximately) to the year.
Comparative genomics
Genome contiguity was assessed by aligning complete virus genomes with NUCmer v3.1, which is a fast aligner for multiple, closely related genomes, using a minimum cluster length of 100 nucleotides. The output was visualized with Mummerplot. 33 To identify orthologous genes, ALRV genomes were first aligned using the Mugsy whole genome aligner (version 1, release 2.3) with default settings 34 and putative orthologs were clustered using Mugsy-Annotator 0.5 35 (coverage cutoff = 0.8, identity = 0.8, and query coverage = 0.8) with GenBank files containing original annotations for each virus supplied as input. Mugsy-Annotator is a command line program, which uses a whole genome multiple alignment and the genomes' associated features to identify annotation anomalies such as fragmentation, inconsistent starts, and missing features. Orthologous genes are grouped in clusters, and the algorithm requires loci to encode functional proteins (ie, no premature stop codons) for cluster membership. The sequence indices generated by Mugsy-Annotator were then used to extract ortholog sequences from whole genomes using the EMBOSS program Extractseq. 36 Sequences were then aligned with reference to translated amino acid sequences using TranslatorX, 37 which was run locally from the command line (with MAFFT as the aligner, amino acid sequences translated from the nucleotide sequences, and default settings used for other parameters).
The amount of variation in amino acid and nucleotide alignments was summarized with AMAS,
38
a python command line utility, which allows the computation of basic statistics from alignments. Alignments were checked for the presence or absence of sequences from each of the 16 isolates to generate a matrix (Supplementary Table 1; 16 viruses and 130 orthologs), which was used to summarize the number of genomes containing the ortholog as well as a summary of its occurrence across the virus phylogeny. In addition, the occurrence of each ortholog in the genomes of the GIV-like ranaviruses was assessed by a BLASTP search (Blast 2.2.29+,
After removing stop codons, orthologs were assessed for recombination with GARD,40,41 which is implemented in HyPhy and uses a genetic algorithm to find recombination breakpoints in multiple sequence alignments. GARD was run in parallel in an MPI environment with five threads using the HKY85 substitution model. Alignments were split at breakpoints identified by GARD for further analysis. Prunier
42
identifies likely occurrences of lateral gene transfer by analyzing the lack of congruence between the topologies of a reference
Each parent alignment and any split alignments (generated with reference to the GARD output using a custom script) were converted to interleaved phylip format using Prank v.140603. 43 These alignments were used to construct phylogenetic trees by maximum likelihood using RAxML 44 ; applying the GTR model of nucleotide substitution with rate variation modeled by a gamma distribution with four categories. Ten maximum-likelihood trees were generated on distinct starting trees in RAxML's rapid hill-climbing mode, and 100 bootstrap replicates were calculated and annotated on the best maximum-likelihood tree.
The adaptive version of Branch-Site REL 45 (implemented in HyPhy) was used to detect episodic selection. Branch-Site REL uses random effects models to allow variation in substitution rates between branches across a phylogeny in order to overcome shortcomings in previous branch-site models of selective pressure that can lead to erroneous inferences. 45 Each alignment was analyzed for positive selection using the random effects branch-site model, applying the universal genetic code and allowing alpha to vary from branch to branch and omega to vary among branch-site combinations. The RAxML trees were provided as input, and all branches were tested before applying the default Holm–Bonferroni correction for multiple comparisons. Positive selection was also analyzed with BUSTED, which uses the Branch-Site REL framework to test for evidence of episodic selection by estimating the proportion of selected codons across all branches of the phylogenetic tree instead of the branch-specific method of Branch-Site REL. 46 Again, the universal genetic code was used.
Phylogenetics
To address the evolutionary relationships among ALRVs, a phylogenetic tree including GIV as an outgroup was constructed. The results of
In order to obtain the maximum amount of data possible for the reconstruction of phylogenetic relationships among ALRVs, nucleotide sequences of the ALRV-core orthologs (only those present in all sixteen ALRVs) without evidence of recombination were used in phylogeny reconstruction. Alignments were concatenated and processed following the same pipeline described above (using 500,000 generations for MCMC chains). This tree was used as the
Results
Genome content
A total of 130 orthologous clusters were found in ALRV genomes. The attempted functional annotation of these orthologs conducted here resulted in only 40 annotations (Supplementary Table 2). The majority of genes were shorter than 1000 nucleotide bases in length (Supplementary Fig. 1). The 26 genes identified previously
29
as core genes that are conserved across the whole family (
Orthologous genes with evidence of recombination.
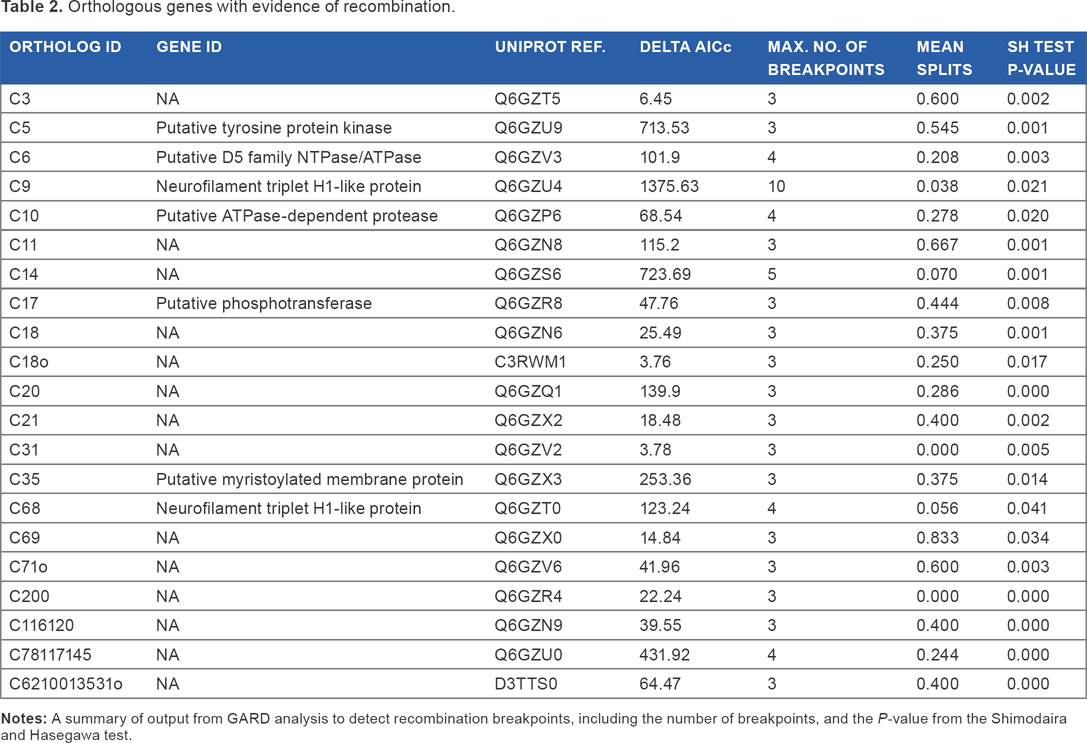
Phylogenetic relationships of amphibian-like ranaviruses (ALRVs) based on 51 ALRV-core genes having removed genes with evidence of recombination. Maximum likelihood and Bayesian trees were constructed from a concatenated nucleotide alignment 44,940 nucleotide bases in length, which was partitioned by loci.
Twenty-nine orthologs were therefore considered clade-specific within the ALRV. Each major clade had four clade-specific orthologs. There were also 12 orthologs in common between the FV3-like and CMTV-like clades that were not found in ATV-likes, and there was one ortholog that was lost in CMTV-likes (ie, present in ATV- and FV3-likes) compared with four orthologs lost in FV3-likes. The rate of functional annotation was very low among these accessory genes: the only orthologs with some available indication of function were a putative integrase-like protein, which was absent in the ATV-likes, a putative capsid maturation protease that occurred only in ATV-likes (and only in the fish pathogens of this group - European sheatfish virus [ESV] and epizootic haematopoietic necrosis virus [EHNV]), and a novel US-22 family protein in the CMTV-likes.
In addition, 38 singletons - defined as novel open reading frames present in only one genome - were found in ALRV genomes. Thirty of these singletons occurred in the two fish pathogens of the ATV-like clade (25 in ESV and five in EHNV), which have larger genomes than the other ALRVs (Table 1). Singletons were also found in ADRV2010 (2), CGSIV (2), CMTV_NL (1), STIV (1), and TFV (2).
Removing genes with signs of recombination resulted in a set of 51 orthologous genes that were found in all 16 ALRVs (ALRV-core; Supplementary Table 2). The final concatenated alignment contained 16 taxa and 44,940 nucleotide bases. Reconstruction of phylogenetic relationships of ALRVs using this 51-gene set resulted in a well-supported phylogeny (Fig. 1). When rooted on ATV-like viruses, the tree returned ATV-like, CMTV-like, and FV3-like viruses as three monophyletic clades with tortoise ranavirus 1 (ToRV1) intermediate to the ATV-likes and CMTV-like/FV3-like clade.
Systematics and genome arrangement
After removing genes with evidence of recombination, a set of 39 orthologous genes present in the GIV-like viruses and all 16 ALRVs (Supplementary Table 2) were retained for use in phylogeny reconstruction. The concatenated nucleotide alignment contained 17 taxa and 39,498 nucleotide bases. The concatenated amino acid alignment contained 13,166 residues. In spite of the large amount of data, phylogenetic evidence for the root of the ALRVs was inconclusive - there was support for ATV on its own or as part of a clade of ATV-like viruses as the root with the topology sensitive to the method followed for tree construction (Supplementary Fig. 2). It was therefore not possible to determine whether ATV-like viruses should be considered monophyletic or paraphyletic.
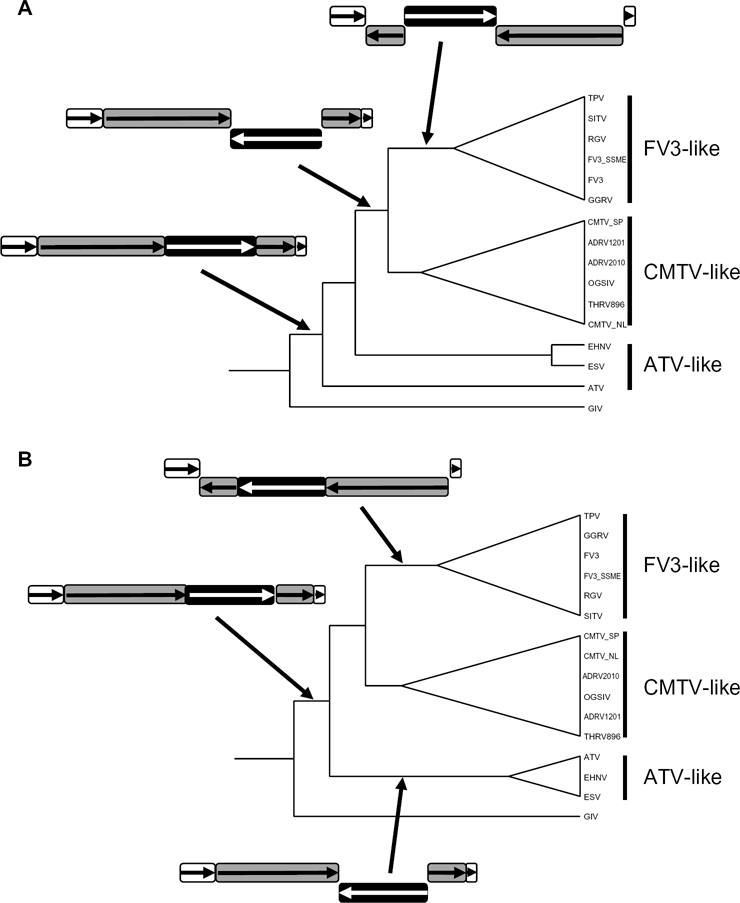
Alternative hypotheses for the evolutionary history of the ALRVs accounting for phylogenetic signal and genome arrangement. Genome segments involved in major rearrangements are color filled: black for the 30 kb rearrangement and the combined black and gray segments for the 88 kb rearrangement (see text). Orientation of genome segments is indicated by arrows and position relative to horizontal axis. (A) Ancestral ATV-like arrangement and paraphyly of ATV-likes. (B) Ancestral cMTV-like arrangement and monophyly of ATV-likes.
An examination of ALRV genome arrangement, using whole genome alignments, did not resolve the ALRV root either. Genome contiguity is very consistent among ALRVs, but there have been two major rearrangements during ALRV evolution (Fig. 2). These rearrangements support the division of the ALRVs into three groups but do not resolve whether there is a systematic basis for all three (ie, groupings which correspond to monophyletic clades). CMTV-like viruses are distinguished from ATV-like viruses by an approximately 30 kb inversion. This inversion bisects an annotated open reading frame at one end in ATV. A steroid oxidoreductase-like protein occurs as a full-length gene in CMTV-like viruses but occurs as a truncated version in ATV (ortholog identifier = C367o). However, this gene is present at full length in EHNV and in the same orientation as CMTV-likes but absent all together in ESV. Therefore, two alternative hypotheses remain concerning the root and ancestral genome arrangement of ALRVs (Fig. 2).
A second major inversion of approximately 88 kb -containing the older 30 kb rearrangement described earlier - occurred on the branch to the ancestor of all FV3-like viruses (Fig. 2). Again, this inversion bisects an open reading frame at one end when the original annotation of the CMTV_SP virus is used as a reference. There were a number of additional smaller rearrangements, the largest an approximately 13 kb long inversion in CGSIV relative to the very closely related ADRV isolates.
Recombination and selection
Twenty-one orthologous genes were found with evidence of recombination (

ToRV1 as a mosaic virus. Colored blocks represent putative genes (the position above or below the line along with the respective arrows indicates orientation). Colors represent ToRV1′s position in the gene tree (within the FV3-like clade, the sister taxon to the FV3-like/cMTV-like clade [root], or unresolved). Tick marks are positioned at 10 kbp intervals.
Twenty-eight genes were found with evidence of episodic selection using the branch-site method (Table 3). Again, functional annotation was lacking, but eight were annotated and three of the 29 were members of the
Amphibian-like ranavirus orthologous genes under positive selection.
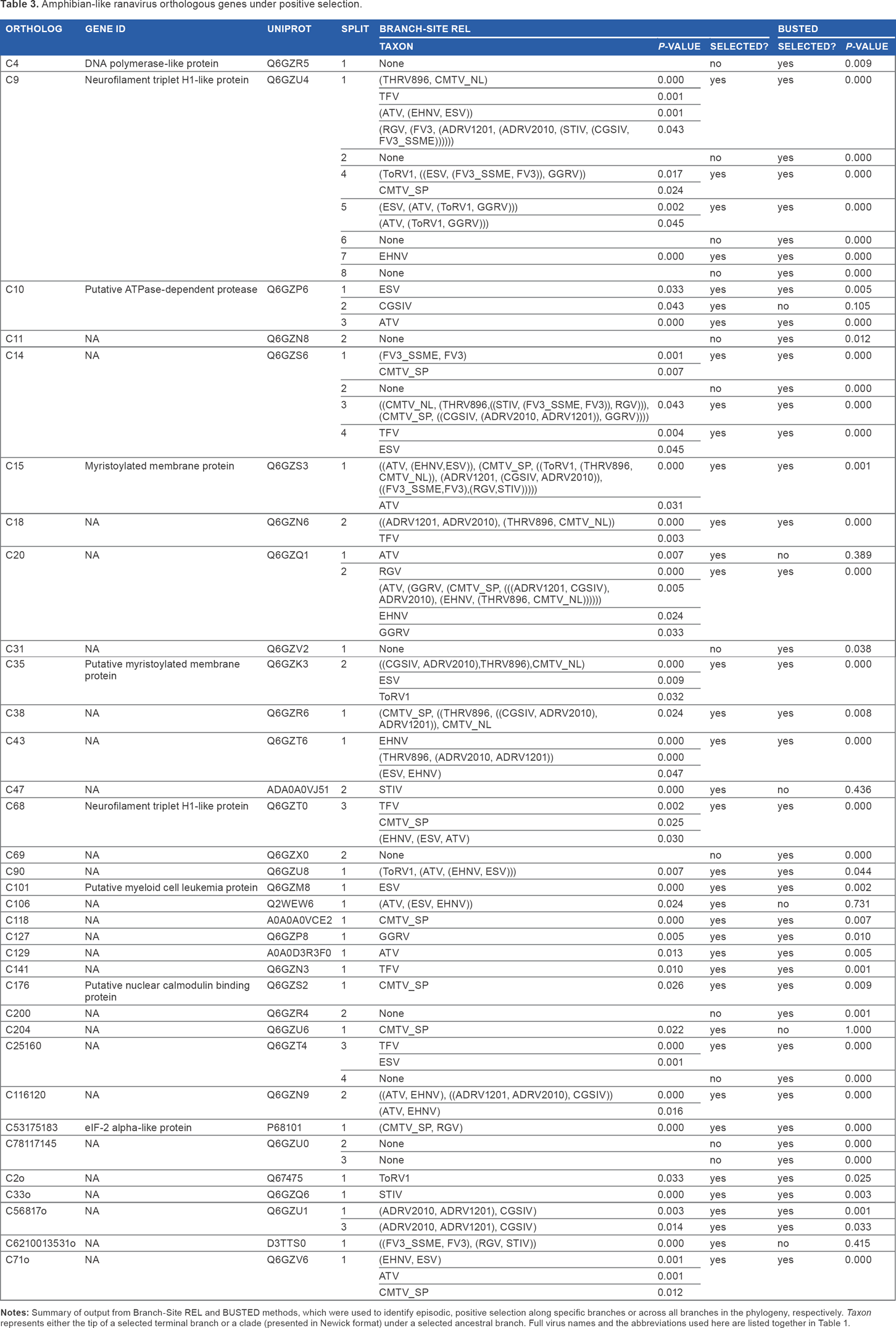
Branch-Site REL identifies the branches in a phylogeny where selection has occurred. Among the 28 genes “with evidence of selection, the terminal branches leading to ATV and ESV (five genes), TFV (six genes), and CMTV_SP (seven genes) featured most frequently. Other terminal branches where positive selection was detected were EHNV (three genes); ToRV1, STIV, and GGRV (all two); CGSIV and RGV (one). In terms of selection on ancestral branches, the ATV-likes and the viruses isolated form Chinese giant salamanders were found within the affected clades most frequently. Annotated orthologs that have experienced selection included a nuclear calmodulin-binding protein gene, neurofilament triplet H1-like protein genes, myristoylated membrane protein genes, and an ATPase-dependent protease.
The alignment-wide approach using BUSTED identified many of the same genes as Branch-Site REL (Table 3): 24 of the 28 genes identified by Branch-Site REL were also significant for positive selection using BUSTED, while there were six additional genes identified by BUSTED as candidates for having undergone episodic positive selection (including DNA polymerase).
Discussion
The traditional view of large DNA viruses was that they were slow evolving and their genomes were lacking in dynamic changes. This study suggests how dynamic genome change in ranaviruses is linked to important aspects of their biology and adds to work on other virus families that challenge this traditional view.9,11 The ALRVs of the family
ALRV genomes were examined for the presence or absence of functional coding sequences to establish core gene sets as well as their accessory genomes. Some of the variation in genome content maps to the virus phylogeny with groups of genes that are lineage specific. ALRVs are highly variable in their host range and virulence,20,21,50,51 and variation in genome content is considered an important factor in explaining observed phenotypic differences between virus types in such key traits.
52
Unfortunately, this study found a similar, low rate of functional information for
Recombination was also found to be widespread in
In addition to assessing orthologs independently for evidence of recombination, the phylogenetic relationships of ToRV1 were compared across its genome. Phylogenetic analysis of each gene along the genome revealed two alternative positions for ToRV1, either at the base of the FV3-like clade or as a sister taxon to the FV3-like/CMTV-like clade. Genes returning each position were spatially clustered, with short sections of the genome supporting one position or the other, suggesting that ToRV1 is a mosaic virus. Of course, alternative explanations could be a sequencing library made up of two divergent viruses arising via lab contamination or coinfection. 55 These alternative hypotheses could be assessed - though not distinguished - with access to the original short reads. This virus was isolated from a captive, exotic reptile originating from the pet trade and as such can be considered at higher risk of exposure to diverse, geographically distant viruses resulting in a genuine mosaic virus or the type of mixed infection that might lead to an error in genome assembly.
There have been a number of attempts to use phylogenetics to address
The 26 genes conserved across the family
The finding that positive selection on
In total, seven genes (more than any other virus studied) were found under selection along the terminal branch, leading to the Spanish isolate of CMTV, which is notable for the observations of its severe impacts on entire amphibian communities. 20 A putative nuclear calmodulin-binding protein was among these. Calmodulin is a calcium receptor that plays a critical role in a variety of cell responses to virus infection. 62 The potential capacity of this protein to modify conserved host pathways makes it an interesting target in efforts to understand the host range of this virus isolate.
As discussed, this study follows a number of others in attempting to use phylogenetics to reconstruct the evolutionary history of ALRVs. Jancovich et al. 19 proposed two alternate patterns of host switching consistent with their analysis of phylogenetic data, and the root of the ALRV was key to distinguish between them. Unfortunately, the analysis presented here failed to identify the root with any confidence since the topology and support was sensitive to the data supplied and the method applied. There was some support for ATV alone as the outgroup (which would mean that ATV-like viruses are paraphyletic) but monophyly of ATV-likes (ATV, EHNV and ESV) was also supported. The monophyly of ATV-likes with ATV at the root of this clade is significant as it suggests that the ancestor of ALRVs was an amphibian pathogen and there was a jump back into fish.
This study also highlighted some interesting results surrounding viruses from aquaculture. There was evidence of frequent positive selection in the tiger frog virus (TFV), which was isolated from an aquaculture facility in China. 53 These findings may therefore support the hypothesis that such facilities provide suitable environments for the rapid evolution of increased virulence. 50 Ranavirus isolates from aquaculture facilities in North America have already been shown to have increased virulence compared to related viruses from the wild, and the potential for spillback of viruses from industry to the wild should be considered a significant conservation threat. 50
Author Contributions
Conceived and designed the experiments: SJP. Analyzed the data: SJP. Wrote the article: SJP. The author reviewed and approved the final article.
Supplementary Material
Footnotes
Acknowledgments
The author thanks Richard Nichols for helpful discussions and also thanks Florent Lasalle for advice and sharing custom scripts to run HYPHY programs in batch. Finally, the author thanks four anonymous reviewers who provided helpful feedback, which improved the article.