
Abstract
This article presents an innovative approach to phylogenies based on the reduction of multistate characters to binary-state characters. We show that the reduction to binary characters’ approach can be applied to both character- and distance-based phylogenies and provides a unifying framework to explain simply and intuitively the similarities and differences between distance- and character-based phylogenies. Building on these results, this article gives a possible explanation on why phylogenetic trees obtained from a distance matrix or a set of characters are often quite reasonable despite lateral transfers of genetic material between taxa. In the presence of lateral transfers, outer planar networks furnish a better description of evolution than phylogenetic trees. We present a polynomial-time reconstruction algorithm for perfect outer planar networks with a fixed number of states, characters, and lateral transfers.
Keywords
Introduction
Among phylogenetic methods, distance-based approaches have gained the somewhat ambiguous reputation to be computationally efficient and to furnish surprisingly good results when compared to the conceptually better character-based approaches. Distance-based approaches are therefore often regarded as a useful evil for example in very large phylogenies when the computational complexity of character-based approaches becomes rapidly so large that even the best heuristics may lead to suboptimal results. In this article, we would like to take a different stand by not merely opposing character- versus distance-based approaches, but by exploring the complementarity of the two approaches. Stevens and Gusfield
1
have shown that any phylogeny on
With the availability of many complete genomes, the importance of lateral transfers in evolution became clearly recognized and increasingly doubts have arisen about the feasibility of drawing a tree of life based on genome evolution.
2
One is today in a somewhat paradoxical situation. On the one hand, almost nobody disputes the existence and importance of lateral transfers in evolution. On the other hand, there are numerous studies that show a good level of consistency between character-based phylogenies and accepted phylogenies obtained with different methods when such phylogenies do exist. An answer to this apparent paradox was suggested for distance-based approaches,
3
namely, that the circular order of the taxa on a tree is quite robust against lateral transfers. The circular order of the taxa on a tree is the order at which the end nodes are encountered in a clockwise scanning of a tree. If lateral transfers are only between consecutive end nodes, the tree reconstructed with the Neighbor-Joining algorithm furnishes a circular order of the nodes.4,5 The order of the nodes corresponds to one of the possible orders of the tree prior to lateral transfers. By using the degrees of freedom on the order of the taxa in a tree, a large number of lateral transfers can be accommodated by a tree while preserving the circular order. To what extent is this result still valid in the case of
In the case of lateral transfers, an outer planar network is a better representation of phylogenetic data than a phylogenetic tree. An outer planar network is a special type of phylogenetic networks that can be reconstructed from a distance matrix using NeighborNet.
6
In distance-based approaches, lateral transfers between consecutive end nodes preserve the circular order of a phylogenetic tree and the distance matrix fulfills the so-called Kalmanson inequalities.
5
If a distance matrix fulfills the Kalmanson inequalities, then the matrix can be exactly described by an outer planar network. The question arises whether there is an efficient method to reconstruct a perfect outer planar network from characters. For a perfect phylogeny, the problem of reconstructing a perfect tree is nondeterministic polynomial time.7,8 If the number of states is a fixed constant
If no outer planar network describes exactly the input data, then one needs methods to judge the quality of a given reconstruction. The reduction to binary characters with missing states allows the introduction of measures characterizing how well a particular phylogenetic tree or outer planar network describes a set of
Phylogenetic Trees and Networks
Phylogenetic methods are divided into two main approaches, character- and distance-based approaches. Character-based approaches are conceptually well suited to evolutionary studies, whereas the low complexity of distance-based approaches permits to deal with a large amount of data. Phylogenetic data are commonly represented under the form of a tree or in the form of a combination of trees as in phylogenetic networks. In this section, the main results that are used in the following sections are presented succinctly, since they have been published elsewhere.1–17 New results are presented in the subsequent section “A common framework for character- and distance-based phylogenies” as well as in Annexes 2 and 3. Annex 1 is a reformulation of known results that is necessary to understand Annex 2.
Distance-based approach to phylogeny
A graph 
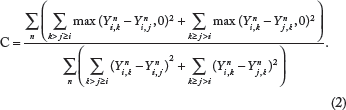
Character-based approach to phylogenetic trees and networks
A phylogeny defined by a set of characters is referred to as a character-based phylogeny. A perfect phylogeny problem has typically as input a character matrix
An unrooted phylogenetic network

A phylogenetic network can be used to represent the effect of a lateral transfer between an origin node and a target node. The lateral transfer may result into a local deviation of a perfect phylogenetic tree represented in the form of a blob.
Reducing a phylogeny on k-state characters to a phylogeny on binary characters
The transformation from multistate to binary characters is done by defining a character Cp, q for each pair (

A
The transformation defined by Eq. (3) applies to any number of character states. For a small number of character states, there is a limited amount of possibilities for completing the missing character states in Eq. (3) to binary characters. For a phylogenetic tree defined by three-state characters, there are three different ways to complete each character;
28
two of them are compatible with the phylogenetic tree. For a set of binary characters, the fulfillment of all four gamete rules is a necessary and sufficient condition for a perfect phylogeny. Is the fulfillment of four gamete rules also a sufficient condition to obtain a perfect phylogeny in binary characters obtained by reduction of
A Common Framework for Character- and Distance-Based Phylogenies
Let us show that a phylogenetic tree defined by a distance matrix can be considered a special case of a phylogenetic tree defined by characters. To do so, the reduction to binary characters approach is extended to evolution models defined by a transition model between bases. Let us start with a simple example: the Jukes–Cantor model. Replacing the missing states ‘?’ in Eq. (3) by 0 leads to the Jukes–Cantor model of DNA evolution. (The Jukes and Cantor
29
model assumes equal base frequencies and mutation rates.) The distance between two taxa is defined as
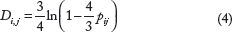
The Kimura two-parameter model distinguishes between transitions and transversions (A or G ⇔ C or T). The Kimura two-parameter distance is given by
30


In summary, the reduction to binary characters’ approach can be applied to both character- and distance-based phylogenies. It provides a unifying framework to explain the differences and similarities between distance- and character-based phylogenies. The main difference between the character-approach and the distance-approach is that in the distance-approach, the transformation given by Eq. (3) does not depend on the set of data. In a distance-based approach, all missing states, ?, are defined for a given model of character evolution. In a character-based approach, a missing state takes a binary value that depends on the input data. It is therefore not surprising that distance-based approaches are computationally less demanding as the search space is much smaller than in character-based approaches. In the reduction to binary characters’ framework, distance-based approaches can be regarded as special cases of character-based approaches. The two approaches are indeed complementary. Character- and distance-based approaches can be combined. Once multistate characters are transformed into binary-state characters, a distance matrix can be computed and a phylogenetic tree or network can be reconstructed using a distance-based approach, such as Neighbor-Joining 31 or NeighborNet. 6 Figure 3 summarizes the last aspect.

The reduction of
How Lateral Transfers Transform a Perfect Phylogeny into a Phylogenetic Network?
Given a tree on multistate characters, a lateral transfer is modeled as an edge (with some direction) from an origin node to a target node on a phylogenetic tree together with possibly a labeling that indicates which characters are transferred. A lateral transfer corresponds to the replacement of some states on the target node by the corresponding character states of the origin node of the transfer. Let us define the target (respectively origin) subtree as the subtree attached to the blob at the target (respectively origin) node. The effect of a unique lateral transfer can be modeled using the assumptions that (i) the target subtree is a perfect phylogeny on the subset of characters defined on the set St of end nodes completed by the target node and (ii) the state of the target node is the only state with possibly one character state common to both the target subtree and the remaining nodes. Figure 4 shows an illustrative example of a lateral transfer. The combined effect of several transfers is described below.

An example showing a split of a perfect phylogeny obtained after removing the target subtree. The lateral transfer preserves the circular consecutive-ones property in the resulting outer planar network obtained after a lateral transfer between adjacent nodes: (0, 0, 1, 1, 1, 1, 1).
In the Jukes–Cantor distance-based model of evolution, the circular order of an outer planar network is quite robust against lateral transfers. One can show that the circular order of the taxa on an outer planar network describing a tree after lateral transfer between consecutive end nodes corresponds to a circular order of the tree.
3
Furthermore, if the tree with lateral transfers is reconstructed with Neighbor-Joining, there is a circular order of the tree that is the same as one of the perfect outer planar network.4,5 To what extent do these results apply to the case of
Proposition 1: If a lateral transfer is between two adjacent nodes on a perfect tree, then there is a circular order of the taxa for which the character states after reduction of the multistate characters to binary-state characters fulfill both the circular consecutive-ones properties and the Kalmanson inequalities and can be represented exactly by an outer planar network.
Proof: The target subtree is a perfect phylogeny on the subset of characters defined on the set St consisting of the target node and the end nodes of the target subtree. Any split in the target subtree is described using Eq. (3) setting all states in S – St to the same binary state as the target state. It is always possible as the state of the target node is the only state with possibly a state common to both the target subtree and the single blob with the removed target subtree. For the remaining pairs of character states, let us set all nodes in the target subtree to the same state. For the rest of the proof, it is therefore sufficient to consider the subtree obtained after removing the target subtree but keeping the target node. Any split in the subtree on the taxa S – St with a splitting vector that is not on the direct path between the origin and the target nodes of the lateral transfer is not affected by the lateral transfer. The only remaining splits that are possibly affected by the lateral transfer are splits with a splitting vector on the path between origin and target nodes. As in this case, all nodes in the target and the origin subtrees have the same state, it follows that a lateral transfer between consecutive nodes preserves the circular consecutive-ones property and consequently the Kalmanson inequalities.
Since for any tree and any two nodes there is always a planar tree representation in which the two nodes are adjacent, 3 the domain of validity of Proposition 1 is quite broad.
Proposition 1 can be generalized to several lateral transfers. In particular, for a level-1 network, the generalization follows from the fact that a level-1 network can be decomposed into single level-1 outer planar networks on which the proof of Proposition 1 applies independently of the other blobs. Let us now consider lateral transfers between adjacent nodes. In order to have a causal model of evolution, one assumes that no edge describing a lateral transfer in a planar representation of the tree crosses another edge and no node is at the origin or target of two lateral transfers.
Proposition 2: If lateral transfers on a perfect phylogeny are such that (i) all lateral transfers are between adjacent nodes, (ii) no edge describing a lateral transfer in a planar representation of the tree crosses another edge, and (iii) no node is at the origin or target of two lateral transfers, then there is a circular order of the tree for which the data fulfill both the circular consecutive-ones properties and can be represented exactly by an outer planar network.
Proof: Given four end nodes in the tree and the shortest paths between the four nodes on the tree, assume that the four nodes have states α, β. A circular order of the four end nodes on the tree with the ordered states α, β, α, β is not allowed on a perfect phylogeny. It follows that such an order may possibly only result from some lateral transfers. This case can also be excluded as lateral transfers are between adjacent nodes, no node is at the origin or target of two lateral transfers preventing crossover, and the target node is the only node with possibly one character state common to both the target subtree and the remaining nodes. Let us show that the reduction to binary states for the pair (α, β) fulfills the circular consecutive-ones properties under a proper choice of the missing states. Consider on the circular order of the taxa the interval between the first node and the last end node with β state, which does not contain the state α, and set all binary states on the interval to zero. The remaining nodes including the nodes with α state are set to one. The reduced states on the circular order of the taxa fulfill by construction of the circular consecutive-ones property.
Proposition 2 shows that given a perfect phylogeny and some lateral transfers between adjacent nodes, the data can be described exactly by an outer planar network on binary-state characters provided some constraints (i–iii) are satisfied. As already known from binary-state characters, a phylogenetic tree constructed with Neighbor-Joining will have a planar tree representation with the same circular order as a perfect outer planar network. 4 In this sense, character-based phylogenies are quite robust against lateral transfers. Contrarily to a perfect phylogeny obtained from a distance matrix, the circular order may not correspond to a circular order of the perfect phylogeny. Quite surprisingly, a phylogenetic tree described exactly by a distance matrix is therefore even more robust against lateral transfer than a character-based phylogeny and may furnish more information on lateral transfers events.
Reconstruction Algorithms for Phylogenetic Trees and Outer Planar Networks
In this section, a reconstruction algorithm for perfect outer planar networks is presented. The main ideas are presented below, although the reader is referred to the Supplementary File for a more detailed description of the algorithm. Let us recall that in a level-1 network, a perfect phylogeny is obtainable by removing at most one edge per blob. A perfect level-1 outer planar network is reconstructed by assembling level-1 phylogenetic networks containing a single blob. The algorithm is an extension of the reconstruction algorithm for perfect phylogenies.
Figure 5B–E illustrates graphically the algorithm. The network is first decomposed into the union of single blob level-1 outer planar networks (Fig. 5B). Each single blob is then decomposed into a blob with single edges attached to it and some phylogenetic subtrees (Fig. 5C). By removing the target node, each blob with single edges transforms into a perfect phylogeny (Fig. 5D) that is reduced into a perfect phylogeny on binary-state characters. The perfect subphylogeny is transformed into a blob by reinserting the taxon at different possible positions within the circular order of the tree and searching for a circular order on which the circular consecutive-ones conditions and consequently all Kalmanson inequalities are fulfilled and the deviation to the four gamete rules is the lowest (It may not be unique.).
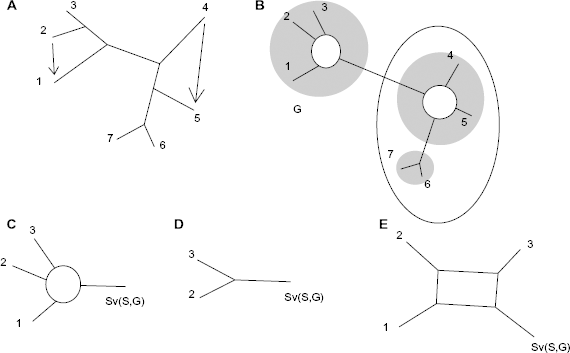
(A) A perfect phylogeny is transformed by lateral transfers between adjacent end nodes into a level-1 outer planar network. (B) The network contains two single blobs. (C and D) Each single blob network is a perfect subphylogeny after removing the target node (there may be several possibilities to remove a taxon and obtain a perfect phylogeny, and in those cases, the algorithm cannot be used to determine the origin and target of the lateral transfer). (E) A single blob description is obtained after reinserting taxon 1.
Dealing with level-1 networks limits the complexity of the reconstruction algorithm as each single blob Nb can be reconstructed independently of the other blobs. The decomposition of a level-1 network into single blobs uses a procedure very similar to the tree decomposition and has a O(23km3n) time-complexity (The higher time-complexity is due to the use of Agarwala and Fernández-Baca's procedure). The phylogeny obtained after removing the target node from a single blob level-1 network with single edges is a caterpillar tree. The target node can be connected to the caterpillar in four different manners. Verifying the circular consecutive-ones condition requires testing four possibilities per blob at the end of the caterpillar tree. The maximum number of blobs is smaller than half the number of taxa. Testing the circular consecutive-ones property has therefore a O(n) complexity. The time-complexity of the algorithm is essentially given by the complexity of reconstructing the single blobs. The time-complexity is quadratic with the number of end nodes. The time-complexity corresponds to the O(22km2n) time-complexity to reconstruct perfect phylogenies multiplied by the number of possibilities to remove a node resulting in a O(22km2n 2 ) time-complexity. The low complexity results from the fact that for each candidate blob with single edges different subsets of taxa are tested, so that considering all blobs, each proper cluster is tested at most 2n+1 times. The second reason for the low complexity is that the fast decomposition procedure 11 can be used here. The perfect reconstruction of a perfect outer planar network has therefore a O(22km2n(n + 2km)) time-complexity.
There is a fundamental difference between level-1 and level-

An example showing how a level-2 outer planar network is decomposed into a level-1 outer planar network: (A) phylogenetic tree with two lateral transfers, (B) single level-2 blob with single edges attached to it, and (C) by removing node 2′, the blob with single edges reduces to a level-1 network.
If the lateral transfer leads to a deviation of the circular consecutive-ones condition, then polynomial-time reconstruction algorithms are not known to us. For level-2 networks, possible lateral transfers explaining the data can be discovered using an algorithm 33 combining the minimum contradiction approach 5 with NeighborNet once the multistate characters are reduced to binary-state characters. As a side comment, let us point out that detecting lateral transfers is important, since a lateral transfer may have a large influence on the apparent rate of evolution if the evolution rate is not constant on all sites.
The perfect reconstruction algorithm for level-1 outer planar networks relies heavily on the reconstruction algorithm for perfect phylogenies, and therefore, the reconstruction algorithm for perfect phylogenies is given in Annex 1 (Supplementary File). In Annex 2 (Supplementary File), the reconstruction algorithm for perfect level-1 outer planar network is presented.
Outlook
Phylogenetic networks are typically used in case of data conflicting with evolution described by a tree. While this article focuses on lateral transfer, let us mention other effects. A crossover between consecutive taxa does not always preserve the circular order. Removing one of the taxa involved in crossover results in a phylogeny fulfilling the Kalmanson inequalities. Discussing gene fusion, complex hybridization schemes, or homologous recombination goes beyond the scope of this article as the effect on a phylogeny will very much depend on how the characters are defined. It could be an interesting research topic.
The extended four gamete rules and the Kalmanson inequalities are two fundamental properties of phylogenies. A perfect phylogeny defined on
Author Contributions
Original idea and first draft: MT. Contributed to the writing of the manuscript: MT, DFB. Agree with manuscript results and conclusions: MT, DFB. Jointly developed the structure and arguments for the paper: MT, DFB. Made critical revisions and approved final version: MT, DFB. Both authors reviewed and approved of the final manuscript.
Supplementary Material
Supplementary Figure 1
The best outer planar network on a dataset for rubber trees from a polymorphic mitochondrial DNA region from rubber tree (
Annex 1
Perfect tree reconstruction algorithms.
Annex 2
Perfect outer planar network reconstruction algorithm.
Annex 3
An example of an outer planar network reconstruction algorithm.
Footnotes
Acknowledgments
We would like to thank the anonymous referees for their pertinent comments and suggestions.