
Abstract
Ergot alkaloids are indole-derived mycotoxins that are important in agriculture and medicine. Ergot alkaloids are produced by a few representatives of two distantly related fungal lineages, the Clavicipitaceae and the Trichocomaceae. Comparison of the ergot alkaloid gene clusters from these two lineages revealed differences in the relative positions and orientations of several genes. The question arose: is ergot alkaloid biosynthetic capability from a common origin? We used a molecular phylogenetic approach to gain insights into the evolution of ergot alkaloid biosynthesis. The 4- γ,γ -dimethylallyltryptophan synthase gene, dmaW, encodes the first step in the pathway. Amino acid sequences deduced from dmaW and homologs were submitted to phylogenetic analysis, and the results indicated that dmaW of Aspergillus fumigatus (mitosporic Trichocomaceae) has the same origin as corresponding genes from clavicipitaceous fungi. Relationships of authentic dmaW genes suggest that they originated from multiple gene duplications with subsequent losses of original or duplicate versions in some lineages.
Introduction
Ergot alkaloids (EA) are a group of mycotoxins causing toxicoses in humans and animals.1–3 Producers of EA include plant pathogens in the genus Claviceps, and some grass endophytes in the genera Epichloë, Neotyphodium and Balansia. 2 These genera belong to the family Clavicipitaceae (order Hypocreales, phylum Ascomycota). EA are also produced by the common airborne fungus Aspergillus fumigatus, a species distantly related with clavicipitaceous fungi, belonging to the family Trichocomaceae (order Eurotiales, phylum Ascomycota). 4 In the same family, some Penicillium species also produce EA.5,6 Relatively few species within these families produce ergot alkaloids and only one representative of the lineages in between the two orders containing these families has been reported to produce ergot alkaloids.7,8 Because of their significant impacts on human health and agriculture, the biochemistry and biosynthesis pathway of ergot alkaloids have attracted much attention and effort. 3 Certain genes involved in the biosynthesis pathway have been characterized.9–15 Clustered arrangements of EA biosynthesis genes have been observed in A. fumigatus, 10 Claviceps fusiformis, 8 and Claviceps purpurea. 14 A gene, dmaW, common to all these clusters, encodes 4- γ,γ -dimethylallyltryptophan (DMATrp) synthase, which has been demonstrated to be the first pathway specific step and has the key regulatory function for EA biosyntheses in Claviceps spp.15–17 Following the cloning of dmaW from C. fusiformis SD58, 18 a gene cluster likely encoding enzymes of EA biosynthesis was identified by sequence analysis in the ergot fungus, C. purpurea P1. 14 This cluster was thereafter designated EAS (ergot alkaloid synthesis). 2 Within the EAS cluster, four genes with known functions were named according to their functions, and the other seven genes with unknown functions were named easA and easC to easH. Homologs of nine EAS-cluster genes have been found in C. fusiformis SD58,18,19 and eight homologs have been found clustered in the A. fumigatus genome 10 (Supplementary Fig. 1).

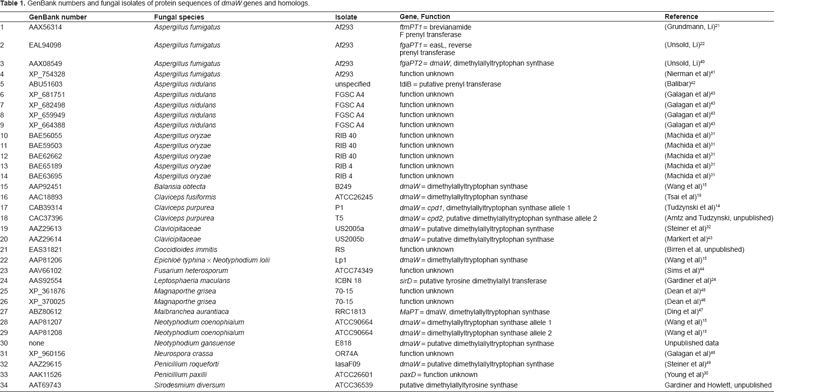
The most parsimonious trees relating amino acid sequences of the deduced products of six dmaW genes and four related genes with known functions. A) Tree based on whole gene region. Of 653 aligned characters, 250 characters are informative. Length = 1521, CI = 0.870, RI = 0.503. Arrows show the discrepancies of the two most parsimonious trees. B and C) Trees based on relatively conserved gene regions screened by GBlocks. Of 308 total aligned characters, 166 characters are informative. Length = 934, CI = 0.864, RI = 0.512. Products of dmaW genes formed a monophyletic group with 100% bootstrap support. B. Unrooted tree; C. Phylogram rooted by choosing the non-dmaW gene products as the outgroup. Numbers on branches indicate bootstrap percentage of MP/posterior probabilities of BI/bootstrap percentage of ML; *indicates that particular branch does not exist in the analysis. The thick branch separates of the outgroup from the ingroup.
Based on the sequence similarity of dmaW genes and general clustering of other hypothetical EAS genes, Coyle and Panaccione 10 proposed that EA biosynthesis in A. fumigatus has a common origin with that in clavicipitaceous fungi. However, arrangement of the cluster genes in A. fumigatus is drastically different from the EAS clusters in C. purpurea and C. fusiformis.2,8 This difference in gene arrangements, and the absence of ergot alkaloids in the lineages between the two groups of EA-producing fungi, raise the question of whether the dmaW genes in clavicipitaceous and trichocomaceous fungi are true orthologs, homologs due to speciation. If the genes are truly orthologous, then multiple gene recombinations and inversions must have happened in either (or both) of the lineages after the divergence from their common origin, and the lineages between them have lost these genes. The alternative would be that the A. fumigatus EAS genes might have evolved independently from different gene duplication events.
Recently, multiple genes similar or related to dmaW have been characterized by cloning and over-expression approaches. Examples include genes for brevianamide F prenyl transferase (FtmPT1) that converts brevianamide to tryprostatin in A. fumigatus, 20 reverse prenyl transferase FGAPT1 catalyzing the final step in the biosynthesis of fumigaclavine C in the same fungus, 21 TdiB with indole alkaloid biosynthetic ability in A. nidulans, 22 as well as SirD, involved in the biosynthesis of an epipolythiodioxopiperazine (ETP) from Leptosphaeria maculans. 23 The available sequences of these genes allow us to look into the evolutionary relationships of dmaW and related genes by a phylogenetic approach.
In this study, we conduct molecular phylogenetic analysis of inferred protein sequences to test whether the known DMATrp synthases in fungi have a single origin or multiple origins. A monophyletic pattern is expected for the single origin hypotheses; whereas a polyphyletic pattern is expected for multiple origins.
Materials and Methods
Protein sequences derived from dmaW genes of clavicipitaceous fungi and A. fumigatus were obtained from previous studies.10,15,18 Protein sequences of FtmPT1, FGAPT1, and the sirD product were downloaded from GenBank (Table 1). The protein sequences from dmaW homologs were obtained by BLAST of the nonredundant protein sequence database in GenBank with the protein product deduced from dmaW from Neotyphodium lolii and A. fumigatus; sequences with relatively high similarity scores and low E-value (<3e−17) were selected (Table 1).
GenBank numbers and fungal isolates of protein sequences of dmaW genes and homologs.
Protein sequence matrix
Due to the high divergence of amino acid sequences, we used the program MAFFT ver.5.8 24 to align them. The alignment was conducted through the web server (http://align.genome.jp/mafft/). The FFT-NS-i and E-INS-i alignment strategies, iterative refinement method, were used to enhance the accuracy of the alignment. The scoring matrix (for amino acid sequences) was selected from six options by comparing fitnesses of the trees in the preliminary analysis. With the scoring matrix selected, we set gap opening penalty (OP = 1.0, 2.0, 3.0) and gap extension penalty (offset value as shown in the program, OF = 0.0, 0.5, 1.0), and again compared the overall fitness of the resulting trees in more preliminary analyses. Parameters resulting in trees of high fitness were set in the final alignment. The alignments by MAFFT were submitted to the program Gblocks 0.91b, 25 to eliminate the highly diverged regions and retain the conserved regions for phylogenetic analysis. Low stringency options were selected to obtain blocks.
Phylogenetic analysis
The protein matrices of ten operational taxonomic units (OTUs, six authentic dmaW and four related genes of known functions) resulting from MAFFT and Gblocks screening and comprised of the conserved regions of the protein alignment, were submitted separately to phylogenetic analysis in PAUP* 4.0b10. 26 Parsimony analyses were conducted using exhaustive search. All characters had equal weight and gaps were treated as missing data.
In order to exclude the possible bias caused by insufficient OTUs, we included multiple potential homologs of dmaW products, which were obtained by BLAST search, in a more extensive phylogenetic analysis. Both conserved protein regions and the whole protein sequence were used in the separate analyses. Parsimony analysis was conducted with a heuristic search with TBR (tree bisection and reconnection) branch-swapping and 100 replicates of random sequence addition. Bootstrapping analysis was based on 1000 replicates of a full heuristic search, each with 20 replicates of a random addition sequence, and tree bisection reconnection (TBR) swapping was selected and re-arrange limit was set to 5000 per replicate.
Bayesian inferences (BI) were performed using MrBayes 3.0B5 27 to analyze individual data sets. The prior for amino acid model was set as mixed to allow model jumping between fixed-rate amino-acid models. Maximum likelihood analysis (ML) was performed using PHYML online server 28 with the following settings: substitution model as JTT, transition/transversion ratio and gamma distribution as estimated by the program, bootstrap datasets 500.
Results
Protein sequence matrix
To choose the appropriate scoring matrix in the amino acid sequence alignment, we compared the overall fitness of the trees based on the alignments of six different scoring matrices in two strategies (Table 2). Setting scoring matrix as JTT200 resulted in the highest consistency index (CI), lowest homoplasy index (HI), relatively high retention index (RI), and shorter trees, thus we choose JTT200 for alignments. In the combination of various OP and OF in the alignment, OP/OF = 1.0/0.0 resulted in three shortest parsimonious trees with a relatively high CI, RI and low HI (Table 3).
Fitness of the trees resulting from maximum parsimony searches with different matrix scoring settings.

Fitness of the trees resulting from maximum parsimony searches with different gap opening penalties (OP) and gap extension penalties (OF) (FFT-NS-i, JTT200).

For the protein sequences derived from six dmaW and four related genes of known functions, an analysis in which the scoring matrix was set as JTT200, OP as 1.0, and OF as 0.0 resulted in an alignment with 653 characters. The resulting alignment was put into Gblocks 0.91b to screen for conserved regions. The number of characters (amino acid positions) retained was 308. For the matrix from the extended OTU set (34 protein sequences), 933 characters resulted from MAFFT alignment, and 155 characters were retained after GBlocks screening.
Phylogenetic relationships
For the data set comprised of ten OTUs, parsimony analyses for the whole regions resulted in two most parsimonious trees (Fig. 1A). The two tree topologies differed in the order of branches ranches to sirD L. maculans and fgaPTl A. fumigatus. The analysis of the conserved regions resulted in one most parsimonious tree. The tree topology differed from those of whole gene regions in the order of divergence of dmaW of A. fumigatus andMalbranchea aurantiaca (Fig. 1B). The known, authentic sequences of DMATrp synthases formed a monophyletic clade with strong bootstrap support. Defining as an outgroup those prenyl transferases known or likely to catalyze production of other products (4 OTUs), the most basal divergence separated the dmaW gene of M. aurantiaca and A. fumigatus from those of the Clavicipitaceae (Fig. 1C).
The analysis with conserved gene regions comprised of 34 OTUs resulted in six equally most parsimonious trees, while the whole gene region (933 characters) resulted in eight equally most parsimonious trees. The variations of branch orders among these trees were mainly from the uncertain positions of the two OTUs, the putative DMATrp synthase gene from Neotyphodium gansuense and paxD Penicillium paxilli AAK11526, which were partial sequences. The strict consensus trees of the six trees from conserved regions and eight trees from the whole gene regions showed the same pattern that, in the dmaW clade, the two putative DMATrp synthase genes from clavicipitaceous endophyte of convolvulaceous plants (AAZ29613, AAZ29614), the clade comprised of C. purpurea and C. fusiformis dmaW, as well as the putative DMATrp synthase gene from N. gansuense collapsed as a polytomy (Fig. 2A). Outside the dmaW clade, paxD P. paxilli AAK11526 along with the other six OTUs appeared as unresolved branches (Fig. 2A). All trees revealed the same monophyletic group of DMATrp synthase genes (functionally tested) and putative DMATrp synthase genes.
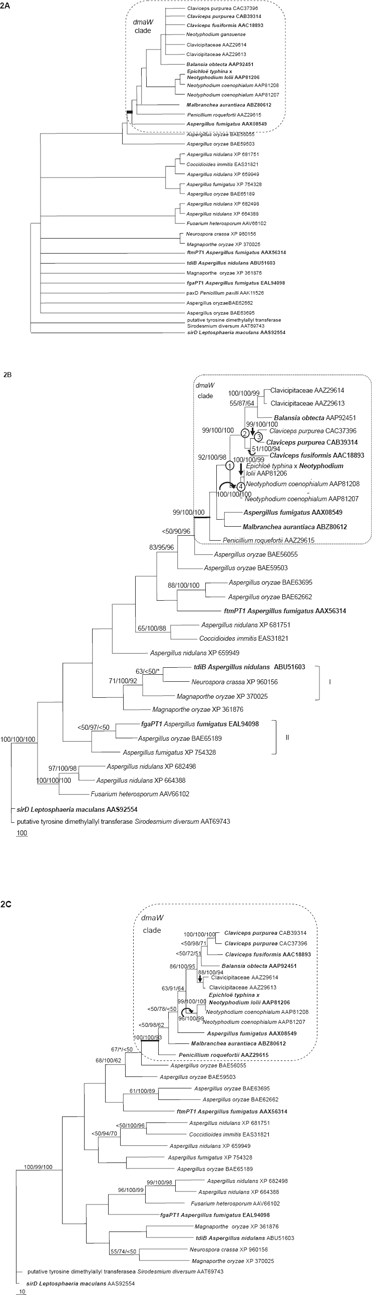
A) strict consensus tree of the six most parsimonious trees for dmaW gene products and products of multiple related genes of 34 OTUs based on relatively conserved gene regions. Of the 155 characters, 133 were parsimony-informative; length = 1366, CI = 0.594, RI = 0.499. B) One of the two most parsimonious trees of 32 OTUs based on the whole gene region. Of the 933 characters, 456 were parsimony-informative characters. Length = 4974, CI = 0.682, RI = 0.499. C) The most parsimonious trees for dmaW gene products and products of multiple related genes of 32 OTUs based on relatively conserved gene regions, which were screened by Gblocks. Total aligned characters = 155, informative characters = 135; length = 1427, CI = 0.607, RI = 0.497. OTUs in bold indicate the gene products with functions that have been confirmed. Numbers on branches indicate bootstrap percentage of MP/posterior probabilities of BI/bootstrap percentage of ML; *indicates that particular branch does not exist in the analysis; numbers in circles indicate the possible gene duplication events (also see Fig. 3).
Excluding the two partial sequences (32 OTUs included in analysis) resulted in better resolutions. Both analyses of conserved regions and the whole gene region resulted in two most parsimonious trees. The two trees from whole gene regions differed in the positions of Magnaporthe oryzae XP 361876, clade I (tdiB Aspergillus nidulans ABU51603, Neurospora crassa XP 960156 and Magnaporthe oryzae XP 370025) and clade II (JgaPTl Aspergillus fumigatus EAL94098, Aspergillus oryzae BAE65189, and Aspergillus fumigatus XP 754328) (Fig. 2B). Comparing the phylogenies inferred from the conserved regions and from the entire sequences, within the dmaW clade, these trees differed in branching orders of B. obtecta, C. purpurea and C. fusiformis dmaW; and in branch orders of A. fumigatus AAX08549, P. roquefortii AAZ29615 and Malbranchea aurantiaca ABZ80612. All clades with strong statistic support (bootstrapping value >70) were present in all trees (Figs. 2B, C), and all trees clearly indicated monophyly of genes for authentic DMATrp synthase.
Bayesian analyses generally resulted in higher statistical support (posterior probabilities) for the clades having high bootstrap support in parsimony analyses (Figs. 1 and 2) and internal branches. ML analyses resulted tree topologies generally congruent with MP tree, i.e. clades with strong support were congruent with MP (Figs. 1 and 2).
All analyses indicated that conserved gene regions selected by GBlocks did not significantly improve the phylogenetic inference for our data sets.
Genes associated with dmaW-related sequences
Certain dmaW-related genes have been identified in gene clusters that are otherwise unrelated to the EAS clusters. These include the sirD gene of L. maculans 23 and the paxD gene of Penicillium paxilli. 29 Because of the close relationship of two A. oryzae homologs, and the availability of a complete genome sequence for that fungus, 30 we checked the genomic contexts of these A. oryzae homologs. Neither of them has a comparable gene cluster. In both cases there are a few apparent secondary metabolism genes nearby, but not homologs of the EAS cluster genes.
Discussion
Various profiles of ergot alkaloids (EA) are produced by EA-producing fungi. Evidence for diversification of EA profiles within an individual fungus, as well as among different producers was observed by Panaccione. 7 The study reported herein is a molecular phylogenetic approach to gain additional insight into the evolution of EA synthesis among fungal producers. Due to its important role in encoding the first step in EA biosynthesis, dmaW was used as a marker to infer the evolutionary relationships of the pathways. Our results demonstrate that dmaW from A. fumigatus and clavicipitaceous fungi formed a monophyletic group indicating that they evolved from a common origin. Therefore we postulate that the EAS gene clusters of the two lineages were also from a common origin, which could be a common gene cluster encoding the shared early steps of the pathways of these two lineages. These shared steps might have been present in the most recent common ancestor of the two fungal lineages.
When 34 OTUs were included in the analysis, a hypothetic gene from P. roqueforti (GenBank accession number: AAZ29615. 31 was grouped in the dmaW clade. Penicillium roqueforti produces the ergot alkaloid isofumigaclavine A, 6 which would require dmaW for its biosynthesis. The product of the P. roqueforti gene was 63% identical with the product of dmaW of A. fumigatus, which was the top match retrieved in a BLAST search with this protein (1e−112). These data are consistent with the P. roqueforti gene encoding the DMATrp synthase that catalyzes the initial prenylation in ergot alkaloid biosynthesis.
GenBank entries often are annotated completely on the basis of BLAST hits. Since the gene from C. fusiformis was identified as encoding DMATrp synthase, 18 many sequences from other species were annotated as putative dimethylallyltryptophan synthase genes according to the similarity of their sequences to the C. fusiformis sequence. However many sequences annotated as dimethylallyltryptophan synthase genes are likely to encode related but nonidentical enzymes catalyzing prenylation or reverse prenylation of different co-substrates or at different positions of the indole rings. Examples of related but functionally different prenyl transferases include the likely tyrosine prenyl transferase in sirodesmin biosynthesis from L. maculans, 23 and the reverse prenyl transferase from A. fumigatus. 21
The rooted tree relating prenyl transferases with known functions showed the most basal separation of DMATrp synthase of A. fumigatus with those of the clavicipitaceous fungi. The EA profile of A. fumigatus includes a series of clavines, simpler tricyclic or tetracyclic alkaloids. In contrast, clavicipitaceous fungi usually produce more complex EA, ergopeptines and other amides of lysergic acid, in addition to the clavines. It is reasonable that more complex functions were gained along the evolutionary path.
Differences in the arrangement of EAS clusters between A. fumigatus and clavicipitaceous fungi were likely caused by multiple gene rearrangements through recombinations, deletions and insertions. The EAS gene cluster of A. fumigatus is in a subtelomeric region. 10 Frequent recombinations associated with such regions provide a potential explanation for the differences between the EAS clusters of A. fumigatus and those of the clavicipitaceous fungi. 7 The chromosomal locations of the clusters in clavicipitaceous species have not yet been determined. Similar rearrangements between more distant genomic locations would account for evolution of clusters, for which selection may favor their inheritance or horizontal transfer as a unit.32,33 Such rearrangements can be driven by repeats such as retroelements in the fungal genomes, such as observed throughout the ergot alkaloid and lolitrem biosynthesis gene clusters in Epichloë festucae and Neotyphodium lolii.34,35
In fungal systematics, the molecular phylogeny indicates that genus Claviceps is more closely related to genus Epichloë (asexual stage: Neotyphodium) than to Balansia;36,37 (also see Fig. 3A). In our 10 OTU gene trees, the clade of C. purpurea and C. fusiformis dmaW was closer to B. obtecta dmaW than to Epichloë spp. dmaW (Fig. 1). The discrepancy between gene tree and species tree can be explained by polymorphic lineage sorting or incomplete sampling. 38 Once we included more OTUs (34 OTUs and 32 OTUs) in the analyses, the dmaW clade separated into two lineages. One lineage was comprised of C. purpurea and C. fusiformis dmaW, the two putative DMATrp synthase genes from clavicipitaceous endophytes of convolvulaceous plants (AAZ29613, AAZ29614), 31 the putative DMATrp synthase gene from N. gansuense, and the DMATrp synthase gene from B. obtecta. The second lineage was comprised of putative DMATrp synthase genes from N. coenophialum and dmaW from the E. typhina × N. lolii hybrid. The divergence of these two lineages and the inconsistency of the divergence pattern with species relationships suggest that dmaW genes in clavicipitaceous fungi have experienced multiple gene duplications and loss of some copies. An alternative is that there may have been some instances of horizontal gene transfer, but the data are not conclusive in this respect. A scenario involving duplications and losses consistent with the evolutionary relationships of authentic dmaW genes is shown in Figure 3B.
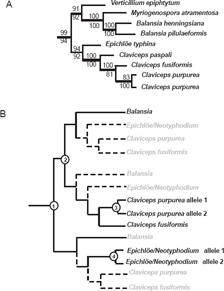
A) Partial phylogenetic relationships of species in Clavicipitaceae inferred by MP based on seven gene regions (Tree redrawn from the data of sung et al). 37 B) Diagram indicating the possible gene duplication events that apparently happened in clavicipitaceous dmaW lineages. Numbers in circles indicate the possible gene duplication events. Dashed lines and shaded OTUs indicate the lost lineages (orthologs).
Disclosure
The authors report no conflicts of interest.
Footnotes
Acknowledgements
This project was supported by the National Research Initiative of the USDA Cooperative State Research, Education and Extension Service, grant number 2008-35318-04549. This article is published with the approval of the Kentucky Agricultural Experiment Station as publication number 09-12-058, and with the approval of the Director of the WV Agricultural and Forestry Experiment Station as Scientific Article number 3037.