
Abstract
Human respiratory syncytial virus (RSV) is a major viral pathogen associated with acute lower respiratory tract infections (ALRTIs) among hospitalized children. In this study, the genetic diversity of the RSV strains was investigated among nasopharyngeal aspirates (NPA) taken from children less than 5 years of age hospitalized with ALRTIs in Hospital Serdang, Malaysia. A total of 165 NPA samples were tested for the presence of RSV and other respiratory viruses from June until December 2009. RSV was found positive in 83 (50%) of the samples using reverse transcription polymerase chain reaction (RT-PCR). Further classification of 67 RSV strains showed that subgroups A and B comprised 11/67 (16.4%) and 56/67 (83.6%) of the strains, respectively. The second hypervariable region at the carboxyl-terminal of the G gene was amplified and sequenced in order to do phylogenetic study. The phylogenetic relationships of the samples were determined separately for subgroups A and B using neighbor joining (NJ), maximum parsimony (MP), and Bayesian inference (BI). Phylogenetic analysis of the 32 sequenced samples showed that all 9 RSV-A strains were clustered within NA1 genotype while the remaining 23 strains of the RSV-B subgroup could be grouped into a clade consisted of strains with 60-nucleotide duplication region. They were further classified into newly discovered BA10 and BA9 genotypes. The present finding suggests the emergence of RSV genotypes of NA1 and BA. This is the first documentation of the phylogenetic relationship and genetic diversity of RSV strains among hospitalized children diagnosed with ALRTI in Serdang, Malaysia.
Keywords
Introduction
The role of the human respiratory syncytial virus (RSV) has been demonstrated as a major viral pathogen associated with acute lower respiratory tract infections (ALRTI) and frequent cause of hospitalization among children less than 5 years.1–4 RSV belongs to the genus Pneumovirus in the family Paramyxoviridae. The genome consists of nonsegmented single-stranded negative sense RNA that encodes 11 separate proteins, 8 of which are structural (L, G, F, N, P, M, M2–1, and SH) and 2 of which are nonstructural (NS1 and NS2, respectively).5,6 RSV was initially divided into 2 distinct major subgroups, A and B, using monoclonal antibodies against virus proteins.7,8 Nucleotide and amino acid (AA) sequence analysis showed that G protein is significantly more divergent than for other RSV proteins for both within and between major groups. 9 Extensive nucleotide and AA variability of the G protein suggests that the virus may reinfect people and cause yearly outbreaks through escaping previously induced immunity.10,11 Extracellular domain of the membrane-bound G protein contains two hypervariable regions separated by a conserved 13 AA motif among all strains. Nucleotide mutations are preferentially accumulated in these two hypervariable regions of the ectodomain. The second hypervariable region of the G protein ectodomain located at the C terminal region provides a reliable surrogate for the variability of the entire G gene. It has been used for phylogenetical and molecular epidemiological studies of the RSV10,12 Being as a target for neutralizing antibody as well as a major constituent for protective antibody is considered another reason for sequence study of the G protein. 13 Phylogenetic analysis using G protein gene revealed that distinct genotypes consist of phyogenetically related strains.10,12 RSV group A has been clustered into 10 genotypes: GA1-GA7, SAA1, NA1, and NA2.10,12,14,15 For the classification of group B, the following genotypes have been proposed: BA1-BA10, GB1-GB4, and SAB1 to SAB3.10,12,13,16,17 Determination of circulation pattern of strains is not easy because of continuous shifting in circulating strains, their multiplicity, and cocirculation during a given season.14,18–20 In recent years, there has been an increasing interest in the genotype study and phylogenetic analysis of RSV strains. However, far too little attention has been paid to phylogenetic study of respiratory viruses in Malaysia. In the current study, we evaluated the genetic variability of the second hypervariable region of G protein gene and deduced AA sequence of both RSV A and B subgroups, detected from hospitalized children with acute lower respiratory tract infections (ALRTI) in Serdang, Malaysia.
Materials and Methods
Clinical samples
Nasopharyngeal aspirates (NPA) were taken from children more than 1 month of age and less than 5 years of age who were admitted from the emergency department to the pediatric wards at Hospital Serdang with the diagnosis of ALRTI within 24 hours of admission from June 16 through December 1, 2009. All potential subjects (including caregivers of the patients) were briefed on the study before written informed consent was obtained. The final selection of the subjects into the study was based on predetermined inclusion and exclusion criteria. Approval from the following authorities was obtained prior to the embarkation of the study: the Medical Research and Ethics committee (MREC) of the Ministry of Health Malaysia and the Medical Research Ethics Committee, University Putra Malaysia. NPAs were transported to the laboratory and refrigerated at 4 °C to 8 °C. In order to avoid repeated freezing and thawing, all NPAs were processed upon receipt. Demographic, clinical features, hospital course, and chest X-ray findings were collected from patients; however, they were not analyzed here.
RSV detection
The D3 Ultra 8 Direct Immunofluorescence Assay (DFA) Respiratory Virus Screening and Identification kit (Diagnostic Hybrids Inc. [DHI], USA) was used as a first step of detection, which made it possible to detect 8 common viruses including RSV, human metapneumovirus (HMPV), influenza virus (IFV) types A and B, Parainfluenzavirus 1–3 (PIV1–3), and human adenovirus (HAdV) using direct immunofluorescence assay (DFA). Positive samples for RSV by DFA were further classified into A and B groups using multiplex reverse transcription polymerase chain reaction (RT-PCR).
RNA extraction and cDNA synthesis
Viral genome was extracted from 0.4 mL of each NPA supernatant by MagMAX Viral RNA Isolation Kit (Applied Biosystems, California, USA) according to the manufacturer's instructions. Reverse transcription was performed on RNA extracts in a final volume of 20 μL by random hexamer primer using RevertAid H Minus First Strand cDNA synthesis kit (Fermentase, Hanover, MD, USA). The samples were incubated first for 5 minutes at 25 °C and followed by 60 minutes at 42 °C. The reaction was terminated by heating at 70 °C for 5 minutes. Seminested multiplex RT-PCR was applied for the detection of a panel of respiratory viruses including RSV, HMPV, IFV types A and B, PIV1–4, and human coronaviruses (HCoV) OC43 and 229E as previously reported. An internal control consisting of glyceraldehydes-3-phosphate dehydrogenase (GAPDH) was included. 21 The presence of human bocavirus (HBoV) and HAdV were individually investigated in the samples by singleplex PCR and nested PCR respectively.22,23 Due to financial constrains, the samples with RSV as a sole agent detected by both methods of DFA and RT-PCR were randomly chosen and proceeded for genotyping study. The second hypervariable region at the carboxy-terminal of G gene of subgroups A and B isolates was amplified and sequenced using primer sets GPA/F1 and GPB/Fl, complementary to the G and F gene nucleotide sequences, respectively. 12 The GPA and GPB primers correspond to the bases 511–530 and 494–515 of the G gene of RSV-A prototype strains A2 and RSV-B prototype strain of 18537, respectively. The F primer corresponds to bases 3–22 of the prototype strains. The PCR was performed in following cycling parameters: 94 °C for 10 minutes denaturation followed by 35 cycles of 94 °C for 50 seconds, annealing at 48 °C for 30 seconds for GPA/F1 and at 52 °C for 30 seconds for GPB/F1 primer sets, 72 °C for 45 seconds, with a final extension of 72 °C for 10 minutes. The PCR products were fractionated by electrophoresis of 2.5% agarose gel and visualized using ethidium bromide under UV light.
Sequencing and alignments
PCR purification was carried when the fragment of expected size was obtained using QIAquick Gel ExtractionKit (QIAGEN, USA), as per manufacturer's instruction. Both strands of PCR products were sequenced using the same forward and reverse RSV primer set with ABI 3730 xl DNA Analyzer (Applied Biosystems) commercially (Repfon Glamor Sdn Bhd, Serdang, Malaysia). Nucleotide sequences of RSV subgroups A and B were assembled separately and manually edited with Bioedite version 7.0.1. Clustal X version 3.06 program of MEGA software, version 4.0 was used for alignment of the sequences. 24 Unique sequences for both A and B subgroup viruses representative of known genotypes obtained from GenBank were included in the phylogenetic analysis. A total of 34 partial sequences (270 nucleotide long) of RSV A and 55 sequences (270 and 330 nucleotide long) of RSV B, previously assigned to specific genotypes, were downloaded from GeneBank for comparative studies. Prototype strains Long (Accession number M17212) for subgroup A and CH18537 (Accession number M17213) for subgroup B were included as outgroups in the phylogenetic analysis.
Phylogenetic analysis
MEGA5 software was used for phylogenetic analyses using maximum parsimony and neighbor joining methods. 25 Genetic distances within/among genotypes were also calculated separately for subgroups A and B based on the sequences used to construct the phylogenetic tree at nucleotide acid level by Kimura's 2-parameter (K2P) models. 26 For Bayesian analysis, Modeltest 3.7 27 was used to determine the optimal model of nucleotide evolution. The (GTR+G) substitution model was selected using an Akaike Information Criterion as implemented in Modeltest. Bayesian Inference (BI) was carried out with MrBayes 3.0 28 to calculate posterior probabilities of recovered clades, with the optimal model of sequence evolution determined from the likelihood ratio test (LRT) s. MrBayes 3.0 was run with 8 × 10 6 generation Markov chain with trees sampled every 100th generation (resulting in 10,000 trees) using default priors. The analyses began on a random starting tree. We discarded the first 5000 trees as a conservative “burn-in,” and the posterior probability values were calculated from the remaining trees. Stationarity was assumed when the cumulative posterior probabilities of all clades were stabilized. A cluster was defined as a bootstrap value of more than 50%. The deduced AA sequences and polymorphisms of the second hypervariable region of subgroup A and B were compared with those of the prototype long strain spanning 270 bp and AY333364 strain (BA strain spanning 330 nucleotide), respectively with MEGA4 software according to the standard genetic codes. The sequences generated in this study have been submitted to GenBank under accession numbers JQ933942-JQ933973.
Results
Occurrence of RSV
A total of 165 children < 5 years of age who fulfilled the inclusion criteria were enrolled and screened for the presence of RSV and other respiratory viruses in the study. RSV was detected in 67 (41%) and 83 (50.3%) of the samples using DFA and RT-PCR, respectively. Using RT-PCR as detection method, RSV was the sole viral agent in 49 (29.7%) of the samples, while 34 (20.6%) samples were coinfected with other viruses including double infections in 29 (17.6%) cases and triple infections in 5 (3.0%) cases. The most prevalent combinations were related to RSV with HAdV (14/34 or 41%) and with HRV (13/34 or 38%). Other viruses including HMPV, HCoV-OC43, IFV-A, and HBoV were codetected in the rest of the samples. RSV positive samples using DFA were further divided into separate antigenic subgroups A and B. Of 67 RSV positive samples with DFA, 56 (83.6%) of the samples categorized as subgroup B and 11 (16.4%), as subgroup A. Thirty-two of 49 samples (9 RSV-A and 23 RSV-B) were RSV was detected as a sole agent were randomly chosen and partial sequences were obtained using GPA/F1 and GPB/F1 primers.
Phylogenetic reconstruction
RSV Group A Strains
The results of model-based methods, Bayesian analyses are largely congruent with NJ and MP analyses for RSV-A and B. We obtained sequence data totaling 270 base pairs for RSV-A and 43 individuals. Of these, 135 characters were constant, and 82 were parsimony informative. MP searches recovered 115 equally most likelihood trees with a consistency index of 0.63 and retention index of 0.87. Phylogenetic analysis showed that all the RSV-A strains were clustered into the recently described NA1 genotype, supported with high posterior probability and high bootstrapping values in maximum parsimony and neighbor-joining (100%, 96% and 98%, respectively) (Fig. 1).

Phylogenese tree for RSV group A nucleotide sequences based on the second variable region of the G protein (270 bp) constructed by the Bayesian analysis method using MrBayes 3.0 software.
RSV subgroup B strains
Relationships of all taxa derived from MP, NJ and partitioned Bayesian analyses of sequences were nearly identical. We obtained sequence data totaling 330 base pairs for RSV-B and 78 individuals. Of these, 151 characters were constant, and 112 were parsimony informative. MP searches recovered 69 equally most likelihood trees with a consistency index of 0.56 and retention index of 0.83.
All the University Putra Malaysia (UPM) strains as well as representative strains with duplicate region were clustered in a clad with bootstrapping value of 86% in a Bayesian analyses. The UPM strains were classified into 3 different genotypes including 19 strains that belonged to the BA10, supported by 100% bootstrapping value, 3 strains belonged to BA9 with 100% bootstrapping value in a Bayesian analysis and one strain that more likely belonged to BAIV genotype (Fig. 2).

Phylogenese tree for RSV group B nucleotide sequences based on the second variable region of the G protein (270 and 330 bp) constructed by the Bayesian analysis method using MrBayes 3.0 software.
Nucleotide and Amino Acid Analysis of Subgroup a Strains
Nine of 11 group A samples were retrieved in sequencing. The average p-distance within the UPM subgroup A strains was 0.01, and it ranged 0.0 to 0.058 within the representatives of other group A genotypes (Table 1). The average intergenotype p-distances between UPM strains and the other genotypes ranged from 0.016 to 0.214 (Table 2). Inferred AA sequences of the second variable region of carboxyl terminal of the G protein for nine RSV-A samples were compared with prototype Long strain (Fig. 3). The results showed that all RSV-A strains included in this analysis exhibit changes in the stop codon position (Gln298 stop) and resulted in predicted G protein of 297 amino acids. Several other AA substitutions occurred in other parts of the studied region is compared with Long strain. AA substitutions specific for NA1 genotype including Asp (D) 237 (except for JQ933945 which was N), Leu (L) 274 (except for JQ933943 which was P), and Ser (S) 292 were demonstrated for these strains. Similar to the GA2 genotype, AA substitutions at Thr (T) 269 and Ser (S) 289, previously assigned as GA2 specific amino acids, were observed in our samples.

Deduced AA alignment of the second hypervariable region of the G protein gene of RSV A for 9 UPM detected strains and 5 retrieved Gene Bank sequences.
RSV-A intragenotype pairwise distances from variation in G gene derived from the Kimura's 2-parameter methods.

RSV-A intergenotype pairwise distances from variation in G gene derived from the Kimura's 2-parameter methods.

Nucleotide and amino acid analysis of group B strains
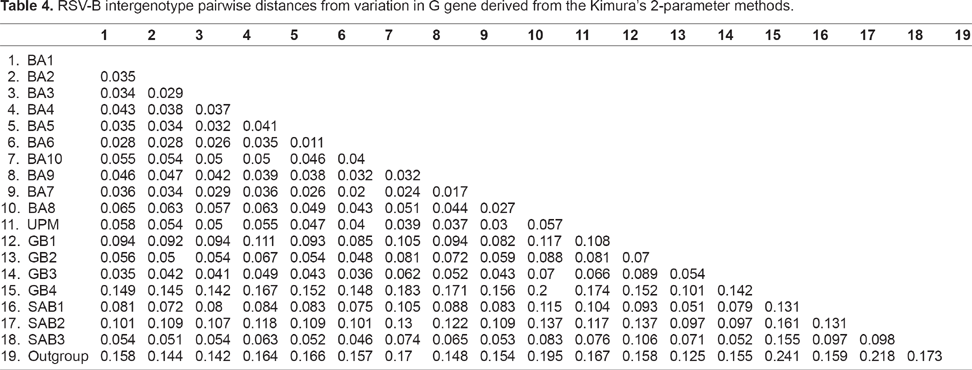
The sequences of the 23 subgroup B strains detected from the patients in this study were included in the phylogenetic analysis. The results showed that all the strains of B A genotypes consisted of a 60-nucleotide duplication region at the second hypervariable region of the G protein gene. The average p-distance within the UPM strains was 0.03 and ranged from 0.002 to 0.042 within the representatives of other group B genotypes (Table 3). The average intergenotype p-distance between UPM strains and other genotypes ranged 0.023 to 0.174 (Table 4). Deduced G protein length and AA alignment of second hypervariable region and stop codons of the strains are shown in Figure 4 for RSV B strains in comparison with the representative sequences. All strains had predicted length of 312 a.a except for 3 sequences (JQ933955, JQ933961, and JQ933962) with predicted length of 319 a.a. In the alignment of deduced a.a sequences with the duplicated segment, AA substitutions were observed in duplicated region as well as in the 60 nucleotide segment exactly upstream. Residue Thr 229 located outside the duplicated region was changed to the He. This change was not observed in other retrieved GenBank strains without duplicated region. All the BA10 strains contained E292G substitution. Strain JQ933961 showed E226D substitution which was not observed in the other BA10 strains.
RSV-B intragenotype pairwise distances from variation in G gene derived from the Kimura's 2-parameter methods.

RSV-B intergenotype pairwise distances from variation in G gene derived from the Kimura's 2-parameter methods.

Deduced AA alignment of the second hypervariable region of the G protein gene of RSV B for 7 representative UPM strains and 5 retrieved GeneBank sequences.
Discussion
The present study was designed to provide a preliminary report on the genetic diversity and molecular epidemiology of RS V detected from children hospitalized with ALRTIs in Malaysia, and to date no similar study has been conducted in this country. Previously published findings have frequently confirmed RSV to be the major viral pathogen associated with ALRTI in children.1,3 RSV was the major reason for hospitalization of children with ALRTI in this study as it represented 50% of the NPA samples tested positive using RT-PCR of which 30% of the cases were single infections. This study is in agreement with previous local studies by Chan 29 and Zamberi, 30 which showed RSV as a major causative agent in Malaysian children. The results further support that RSV infection is a frequent cause of hospitalization among children in tropical and developing countries. 4 In the majority of the RSV studies in developing countries, the prevalence of RSV associated with ALRTI was underestimated due to the application of less sensitive detection methods such as ELISA or immunofluorescence assays. 31 During the 6-month study from May to November, a continuous activity of RSV was seen with a small peak in July, and then RSV activity started to increase from August to October with a relatively big peak in October. The finding seems to be consistent with RSV outbreaks during the wet season in tropical and subtropical countries with seasonal rainfall. 32 However, this study only presents a small picture of the circulation pattern of the RSV genotypes in Malaysia. For a more representative result, a study spanning several years is required since variation in prevalence from year to year is common in respiratory infections.
RSV strains were further classified into A and B using RT-PCR and further sequencing of the partial G protein. During the study period, both antigenic subgroups of A and B cocirculated with predominance of subgroup B. These results differ from some published research that shows the predominance of subgroup A virus among the strains.19,20 However the results of these studies are consistent with those of other published studies that show RSV-B as the dominant group. On the other hand, Gilca (2006) also found rapid changes from A to B during 2 consecutive seasons. This suggests that there is a variation in the pattern of RSV circulation in different regions and seasons.
RSV subgroup B viruses are generally divided into the genotypes comprising nonduplicated regions (GB1 -4 and SAB 1–3) and genotypes with a duplicated segment namely BA genotype. Since the report of BA genotypes with 60-nucleotide insertions in the second hypervariable region of G gene by Trento, 17 the partial sequences of this genotype has been reported worldwide.19,33–36 Results of different tree methods for RSV-B confirmed the K2P distance method and defined that the least mean intergenotype distance was found between genotypes with duplicated regions and UPM strains and the most mean intergenotype distance belonged to genotypes that were not duplicated. The detection of the BA strains in this study further supports the widespread dissemination of the BA strains in the community. It was speculated that all B A genotypes with duplicated regions shared a common ancestor that was separate from other group B genotypes. 13 It was classified into BA-I to BA-VI using G protein gene analysis. Rapid generation of the genetic variation among BA strains is partly related to the immunological selection at duplicated regions in the ectodomain of the G protein. The fast dissemination of genotypes with duplicated regions may be related to the fact that the human immune system may not be exposed to these new genotypes. 13
In a detailed phylogenetic analysis of BA strains by Dapat in Japan, 37 additional new BA genotypes including BA7, BA8, BA9, and BA10 were introduced that were previously described as BA4 genotypes. These newly introduced genotypes comprised the majority of the RSV strains from 2005 to 2010. The different methods of phylogenetic trees largely supported that UPM B strains were separated into 2 different clades, namely BA9 and BA10 genotypes. These findings of the current study are consistent with the Dapat study, which found cocirculation of several BA genotypes in all studied periods with domination of 1 genotype in each season. The present finding seems to be also consistent with other research that found BA genotypes are replacing the previously dominant RSV B genotypes.13,35 Among all RSV-B sequenced strains, residue Thr229, located outside the duplicated portion was changed to the He. This change was not observed in other retrieved GenBank strains without duplicated regions. This finding is in agreement with Trento et al 13 who showed that T229I change occurred in all BA strains with duplicated regions. Consistence with that study, the S247P AA substitution was found in all B strains. The strains JQ933971, JQ933972, and JQ933973, which were classified in clad-included BA9 strains, had a substitution of V271A. Except for strain JQ933953, the rest of the strains clustered in the BA10 had a E292G change. Also E226D change was seen in JQ933961. The results are consistent with the study by Dapat. 37 However, other changes including T289I and S269P were not observed in this study. G protein length diversity due to the position of the final stop codon was observed among RSV B strains, which leads to a deduced G protein length of 312 or 319 amino acids.
The novel emerging of NA1 and NA2 genotypes was reported in 2009. 14 These 2 recently reported genotypes were recognized as variants of GA2. In spite of genetic relatedness between these novel genotypes, they are different antigenically 14 Different methods of phylogenetic tree (BI, MP, and NJ) suggested that the RSV-A strains detected in this study were clustered within the NA1 genotype. There is remarkably good agreement between the result of pairwise distance methods and molecular trees of different methods (BI, MP, and NJ). There is a remarkable match between the molecular phylogeny and genetic distance among UPM subgroup A and NA1 genotype in this study. However, all RSV-A strains in this study showed a 297 AA in length.
Circulation of different genotypes of RSV groups A and B in the study emphasize further epidemiological and clinical surveillance of RS Vin Malaysia. Host immune system evading and reinfections by RSV could be due to the within and between subgroup antigenic variations. 14 The presence of emerging viruses and the lack of relative protective immunity to the new strains may facilitate effective spread of these new strains. 12 The variation in the G protein length may contribute to the antigenic changes and escape from the immune system. 38 However, further studies are required to determine the role of the G protein length on RSV immunity. The findings obtained in this study have important implications for the understanding of the evolutionary pattern of RSV as well as vaccine development. Previous studies have shown that genotype shifting during consecutive years is a common feature of the molecular epidemiology of RSV infections. 12 However, caution must be taken as study at multiple locations and for several consecutive years with a large sample size is required to better appreciate the epidemiology of the RSV. Long-term epidemiological surveillance of RSV strains is important to illuminate antigenic and genetic bases of the infections by RSV.
In conclusion, our study is the first report of the genetic diversity of the RSV strains according to the G gene second hypervariable region during a period of 7 months in Malaysia. Both group A and B subgroups cocirculated in the studied patients, although group B dominated. The BA strains, with 60-nucleotide duplicated region, were identified as the dominant genotype in the samples. The majority of BA strains comprised recently emerging BA10 genotype. However, further long-term surveillance during several epidemic seasons are required to understand the genetic variability and circulation pattern of the RSV strains in the Malaysia.
Author Contributions
MRE performed laboratory methods, analyzed the data and wrote the first draft of the manuscript. ZS as a corresponding author designed the study and verified all the data. NO was involved in obtaining ethics approval from Hospital Serdang and the Ministry of Health Malaysia and conducted the study design. MSL conducted study design. FYM involved in phylogenetic analysis design and data interpretation. All authors have read and approved the final manuscript.
Competing Interests
Author(s) disclose no potential conflicts of interest.
Disclosures and Ethics
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.
Footnotes
Acknowledgment and Funding
This work was partially supported by grants from the Ministry of Science, Technology and Innovation Malaysia (grant number 5450401). We are grateful to all the doctors and nurses of Pediatrics Department, Hospital Serdang for providing nasopharyngeal aspirates for the study.