
Abstract
Pulmonary arterial hypertension (PAH) is a chronic and progressive disease with a poor prognosis if left untreated. Pathophysiological alterations in this disease lead to vasoconstriction, endothelial and smooth muscle cell proliferation, and in situ thrombosis. Endothelin-1 (one of the most potent vasconstrictors known), has been shown to be increased in PAH, contributing, in part at least, to these abnormalities. Endothelin acts through the binding of two receptors, ETA and ETB. Sitaxentan is a selective ETA endothelin receptor antagonist that has been demonstrated, in several clinical trials, to improve exercise capacity, functional class and hemodynamics. Sitaxentan has a good safety profile, is well tolerated and has a low incidence of liver toxicity.
Introduction
Pulmonary arterial hypertension (PAH) is a chronic and devastating condition characterized by a progressive increase in pulmonary vascular resistance (PVR) that ultimately produces right heart failure and death.1,2 This increment in PVR is produced by endothelial dysfunction, abnormal proliferation of the smooth muscle cell and in situ thrombosis.1,2 The term PAH includes a group of diseases that share some pathophysiological mechanisms that make them suitable to be treated with the same drugs.3,4 These diseases are: idiopathic PAH; hereditary PAH; PAH induced by drugs or toxins; and PAH associated with systemic-to-pulmonary shunts, collagen vascular disease, portal hypertension, human immunodeficiency virus, schistosomiasis and chronic hemolytic anemia (group 1 of the Dana Point pulmonary hypertension classification).3,4 PAH is still an incurable disease; however, with the rapid advance in the knowledge of the main altered pathways responsible for its development and progression (endothelin, nitric oxide and prostacyclin pathways), 2 new compounds to treat this disease have been made available in recent years. Sitaxentan sodium, one of these new drugs, is a highly selective endothelin A receptor antagonist (ETA) that has proven effective for the treatment of patients with PAH, improving exercise capacity, World Health Organization functional class (WHO FC; Table 1) and hemodynamics. In this paper, we will discuss the pharmacology, evidence for and indications of the use of sitaxentan in the treatment of PAH. 5
WHO functional classification for pulmonary arterial hypertension.
The Endothelin System
Endothelin (ET) was first isolated from porcine endothelial cells in 1988. 6 Since then, the ET system has been found to be involved in multiple physiologic functions related to the nervous, renal, cardiovascular, respiratory, gastrointestinal and endocrine systems.7–11 The endothelins are a family of isopeptides named ET-1, ET-2 and ET-3, where ET-1 is the most abundant and clinically important. ET is synthesized from preproendothelin, a 212-amino acid peptide, and subsequently cleaved to yield a 38-amino acid propeptide, big ET. Big ET is converted to the 21-amino acid peptide ET by the endothelin-converting enzyme.12–14 ET is produced mainly in the vascular endothelial cells and, to a lesser extent, in pulmonary smooth muscle cells and fibroblasts. 12 A variety of factors can stimulate the production of ET, including hypoxia, ischemia, angiotensin II, vasopressin, catecholamines, cytokines, growth factors and thrombin. 15 ET is one of the most potent vasoconstrictors known, but also has proliferative effects and has been implicated in many disease states including carcinogenesis, bronchoconstriction, fibrosis, heart failure and pulmonary hypertension. 7 A great amount of evidence has shown the important role that the ET system plays in the pathophysiology of PAH: patients with PAH have increased levels of ET in small pulmonary arteries, 16 and increased levels of ET correlate with the severity of disease in adults with idiopathic primary PAH. A strong correlation was found between increased plasma concentrations of ET and increased pulmonary vascular resistance, as well as increased mean pulmonary arterial pressure, in a small cohort of subjects with primary pulmonary hypertension. In addition, a decreased six-minute walk distance (6MWD) correlated with ET levels. 17 Once activated, ET has a dual paracrine and autocrine role: 15 it binds to two receptors known as endothelin receptor A (ETA) and endothelin receptor B (ETB). ETA and ETB are expressed on smooth muscle cells and cardiac myocites; however, only ETB receptors are expressed in the endothelial cells. 13 Activation of both ETA and ETB receptors in the pulmonary smooth muscle cell produces vasoconstriction and proliferation, while the activation of ETB receptors in the endothelial cell mediates ET clearance, and regulates vascular tone through the production of nitric oxide and prostacyclin.13,15 Two types of endothelin receptor antagonists (ERAs) have been developed for the treatment of PAH, one nonselective ERA (bosentan) and two selective ETA receptor antagonists (sitaxentan and ambrisentan). 13 The rationale for using an ETA-selective antagonist is based on the evidence that the ETB receptor is involved in endothelin clearance from the blood, so blocking this receptor could increase circulating ET. Also, blockade of endothelial luminal side ETB receptor might theoretically decrease nitric oxide and prostacyclin production, favoring vasoconstriction.13,15,18
Sitaxentan is an orally active competitive ERA that has more selectivity for the ETA receptor.5,19 In vitro studies have shown that sitaxentan binds almost completely to the ETA receptor, and its ETA:ETB affinity ratio is about 6000:1 (compared to bosentan (40:1) or ambrisentan (77:1)). 15 Sitaxentan decreases mean pulmonary arterial pressure (mPAP), pulmonary vascular resistance (PVR) and right atrial pressure (RAP), with no effect on heart rate, mean arterial pressure, capillary wedge pressure, cardiac index or systemic vascular resistance during intravenous infusion. 5 In addition, sitaxentan reversed and prevented vascular remodeling in animal studies.5,15 Sitaxentan is orally active with a bioavailability of 89% and reaches maximum plasma concentrations 1–4 hours after administration. 5 It is highly bound to plasma proteins and metabolizes in the liver through cytochrome P450 (CYP)2C9 and CYP3A4 isoenzymes. Sitaxentan is a weak inhibitor of CYP2C19 and CYP3A4, and is a moderate-to-strong inhibitor of CYP2C9. The interaction with CYP2C9 causes inhibition of warfarin metabolism, a clinically frequent interaction, considering the need for anticoagulation in patients with PAH. Therefore, dosing adjustments may be necessary. It is recommended to decrease the warfarin dose by 80%, with subsequent dose tritation as needed. 5 Approximately 50%–60% of a single sitaxentan dose is excreted as metabolites in the urine.5,15
Clinical Evidence for the Use of Sitaxentan in PAH
Barst et al 20 published the result of an open-label pilot study to evaluate the safety and efficacy of the oral administration of sitaxentan sodium in patients with PAH (Study 211). It enrolled 14 adults and 6 children with idiopathic PAH and PAH associated with systemic-to-pulmonary shunts or collagen vascular disease, WHO FC II to IV. The sitaxentan dose range was established according to body weight between 100 mg and 500 mg twice a day (6 mg/kg BID) for 12 weeks. 6MWD tests and right heart catheterization were performed at baseline and after 12 weeks. All patients showed a significant improvement in exercise capacity (37 ± 60 m; P = 0.01) and hemodynamics (a decrease in mPAP and PVRi). Serious adverse events included two cases of acute hepatitis (fatal in one patient) that developed in the extension phase of the study; after these events, all patients were discontinued from the extension study without further complications. The cases of acute hepatitis were ultimately attributed to the high dose that patients received and to the non-linear pharmacokinetics observed in the study.
The Sitaxentan To Relieve ImpaireD Exercise (STRIDE) program was developed to evaluate the safety and efficacy of sitaxentan in patients with PAH and included three randomized placebo controlled trials (STRIDE-1, STRIDE-2 and STRIDE-4), one non-controlled study (STRIDE-6) and three long-term studies (STRIDE-1X, STRIDE-2X and STRIDE-3). 18
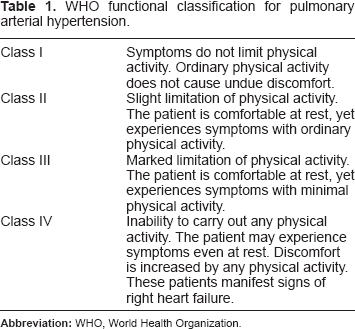
STRIDE-1 21 was a randomized, double-blind, placebo-controlled study that enrolled 178 patients with PAH at WHO FC II to IV. The study was carried out in 23 centers in USA and Canada to evaluate the safety and efficacy of two different doses of sitaxentan (100 mg and 300 mg once a day (OD)) for 12 weeks. The primary end-point was change in peak oxygen consumption (VO2) at Week 12 evaluated via cardiopulmonary exercise testing (CPET). Secondary end-points included 6MWD, functional class, VO2 at anaerobic threshold, VE per carbon dioxide production at anaerobic threshold, hemodynamics, quality of life and time to clinical worsening. Only the 300 mg group increased peak VO2 as compared to the placebo group (+3.1%; P < 0.01). The other endpoints derived from the cardiopulmonary exercise testing were not met. However, both the 100 mg and 300 mg dose significantly increased 6MWD test results (Fig. 1), and also improved functional class and cardiac index, and decreased indexed PVR (PVRi). The incidence of increased liver enzymes was greater in the 300 mg group than the 100 mg group (10% vs. 3%), and the incidence of serious adverse events was infrequent, with no significant differences among treatment groups (placebo 15%, 100 mg 5%, 300 mg 16%). There was one death in the 300 mg group that was ascribed to aggravation of PAH and not linked to the STRIDE-1study. While the overall results of this study were promising, the primary end-point was not met and this may have obscured the impact of the efficacy results. 5 The discrepancy between the results of the CPET and the 6MWD test was unexpected and raised concerns about the reliability of CPET and 6MWD as primary end-points. 5 Oudiz et al 22 showed that while the intracenter correlation of change in CPET and 6MWD was reliable, the inter-center results were not comparable. This was partly due to the lack of CPET and 6MWD validation protocols. Also, the population included in STRIDE-1 was, at that time, somehow different from previous multicenter controlled trials of PAH. Functional class II was included and there were no “upper limit” exclusion criteria for the 6MWD at baseline. It has recently been showed that the 6MWD is limited by a point where performance is so good or so bad that further clinically and/or statistically significant deterioration or improvement is hard to detect (a “floor or ceiling effect”).5,23 Langleben et al 24 performed a post hoc analysis in a subset of patients included in STRIDE-1 that met the traditional criteria for PAH studies (functional class III/IV and 6MWD < 450 m) comparing exercise capacity and hemodynamics at baseline and after 12 weeks of treatment, and found a statistically significant treatment effect in the treated group compared to the placebo group in 6MWD (39 ± 10.65 vs. 26 ± 13.39 m); right atrial pressure (-1.2 ± 0.5 vs. 2.1 ± 0.8 mmHg); mean pulmonary arterial pressure (-4.7 ± 1.5 vs. 0.4 ± 0.5 mmHg); cardiac index (0.38 ± 0.06 vs. −0.09 ± 0.09 L/min/m2) and pulmonary vascular resistance (-274 ± 47 vs. 85 ± 60 dyne/s/cm5). He found also an improvement in functional class (P < 0.0005).
STRIDE-1. Mean change in 6MWD from baseline to Week 6 and Week 12 in placebo and sitaxentan groups; P < 0.01 for the comparison between each sitaxentan dose and the placebo. Data from Barst et al. 21
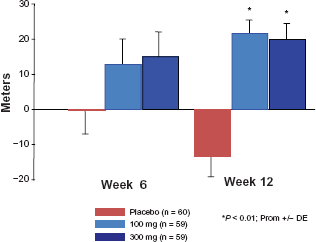
STRIDE-2, 25 a 18-week multicenter randomized placebo-controlled trial, compared two doses of sitaxentan (50 mg and 100 mg, OD) versus a placebo. Also, an open-label bosentan arm (the only oral therapy approved for the treatment of PAH at that time) was included. Two hundred and forty-seven PAH patients, with idiopathic PAH or PAH associated with connective tissue disease or congenital heart disease, were enrolled. The primary end-point was a change in 6MWD from the baseline to Week 18. Secondary end-points included change in WHO FC, time to clinical worsening and change in Borg dyspnea score. Patients treated with 100 mg sitaxentan had an increased in 6MWD compared to the placebo group (treatment effect of 31.4 m; P = 0.03) (Fig. 2) and improved WHO FC (P = 0.04). The incidence of elevated hepatic transaminases was 6% for the placebo, 5% for 50 mg sitaxentan, 3% for 100 mg sitaxentan and 11% for bosentan. Sitaxentan performed in a similar manner to bosentan, which was included in the study to allow a qualitative comparison in a similarly randomized population.
STRIDE-2. Mean change in 6MWD from baseline to Week 18 in the placebo, 50 mg sitaxentan, 100 mg sitaxentan and bosentan groups. There was a significant improvement in 6MWD in the 100 mg sitaxentan group and the bosentan group compared with the placebo group. Modified from Barst RJ, et al. 25
STRIDE-4 (unpublished) was a multicenter, placebo-controlled study that was conducted in Latin America and Europe, its design was the same as that of STRIDE-2 except for the open-label bosentan arm. Ninety patients were enrolled. Sitaxsentan at a dosage of 100 mg improved functional class in PAH patients, who were mostly WHO FC II at baseline. No patient receiving 100 mg sitaxsentan experienced clinical worsening. The primary end-point (imrovement in 6MWD) was not met, probably because this study was not able to account adequately for the large placebo effect observed and the high proportion of funcitonal class II patients included (61%). The safety profile was similar to other STRIDE studies.
STRIDE-6, 26 a double-blind exploratory study, was completed in 48 PAH patients discontinuing bosentan because of safety issues or lack of efficacy These patients were randomized to receive sitaxentan at doses of 50 mg or 100 mg OD for 12 weeks. Thirty-three percent of patients receiving the 100 mg dose and 10% of patients in the 50 mg group improved their 6MWD by more than 15%, while 20% and 15% of these patients had a >15% decrease in 6MWD in the 100 mg and 50 mg groups, respectively. Of the 12 patients discontinuing bosentan because of hepatotoxicity, only one had elevated liver enzymes at 13 weeks after sitaxentan therapy. From these data, it appears that sitaxentan may represent a safe and efficacious alternative in patients discontinuing bosentan.
Only the results from two extension studies (STRIDE-1X and STRIDE-2X)27,28 to evaluate the long-term efficacy and safety of sitaxentan have been reported. In STRIDE-1X, 27 11 patients that completed the placebo-controlled 12-week study were allowed to continue in a extension phase receiving either 100 mg or 300 mg sitaxentan OD. 6MWD, functional class and hemodynamics were assessed before sitaxentan was initiated and after one year of drug therapy. After one year of sitaxentan use, 6MWD increased by 50 m (P = 0.04) and functional class improved (before therapy, nine patients were in WHO FC III and one patient was in WHO FC II; at follow-up, all patients were in FC II (P < 0.01). Although the mPAP did not change, cardiac index and PVR improved significantly There were no serious adverse events and, notably, no elevations of hepatic enzymes. STRIDE-2X 28 was an international open-label study that evaluated all-cause mortality, time to discontinuation (all causes) from monotherapy, time to discontinuation due to adverse events, time to elevation in hepatic enzymes, time to discontinuation due to elevated hepatic enzymes and time to first clinical worsening in the group of patients receiving 100 mg sitaxentan (145 patients) and bosentan (84 patients) at one year. Patients that were initially randomized (STRIDE-2) to the 50 mg group continued receiving 100 mg sitaxentan during the open-label phase but were excluded from the final analysis. At one year, patients treated with 100 mg sitaxentan OD had 96% overall survival and a 34% risk for clinical worsening. There was a 6% risk of elevated transaminases (>3x upper level of normal (ULN)) and a 15% risk of discontinuation due to adverse events. Patients receiving bosentan had 88% overall survival and a 40% risk of clinical worsening at one year. In addition, there was a 14% greater risk for elevated hepatic enzymes (>3x ULN) and 30% discontinuation due to adverse events. Interpretation of the results of STRIDE-2X must be made with caution, as the authors point out, because of the lack of blinding of the sitaxentan and bosentan groups. Furthermore, the one-year survival estimates for sitaxentan (96%) and bosentan (88%) reflect the initiation with ERA monotherapy for the treatment of PAH but are not necessarily attributable to the ERA therapy alone. 28 These two long-term studies support the findings of prior studies evaluating sitaxentan with respect to safety and efficacy, and they also provide support for durability of efficacy.
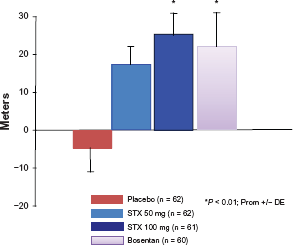
Our group presented the results of a three-year follow-up of 36 patients with PAH, functional class II and III, receiving 100 mg sitaxentan OD. Cumulative survival at one, two and three years was 96%, 79% and 75% respectively (Fig. 3). Five patients required combination therapy (sildenafil) because of clinical deterioration. 29
Survival estimates at five years from sitaxentan initiation (n = 55). From Pulido et al. 29
We can conclude that sitaxentan significantly improved functional class (STRIDE-1, STRIDE-2, STRIDE-4), 6MWD (STRIDE-1 and STRIDE-2), dyspnea score (STRIDE-1) and hemodynamics (Study 211 and STRIDE-1). Improvements in time to clinical worsening could only be demonstrated in a post hoc analysis using the data from the three pivotal studies. 18
Safety
The most frequent adverse events with sitaxentan treatment include headache, peripheral edema, nausea and nasal congestion, which are well tolerated. The most important adverse event, however, is the increase in hepatic transaminases. This abnormality appears to be a class effect of ERA therapy and has also been reported with bosentan and, less frequently, with ambrisentan. 5 Abbott et al 30 found an average increase in transaminases of 4% after four different clinical trials were analyzed. In these studies, sitaxentan exposure ranged from 12 to 28 weeks. Longer studies have shown a one-year risk of elevation of hepatic transaminases (>3x ULN) of less than 4% (Kaplan–Meier analysis) at a dose of 100 mg OD, which is lower than that reported with bosentan (14%–15%).27,28
To our knowledge, five patients have developed acute hepatitis and liver failure while receiving sitaxentan.20,31–33 The first two cases (one of them fatal) received sitaxentan at a dose of 300 mg OD, which is much higher than the presently approved dosage of 100 mg. 20 The second report 31 was that of a 25-year-old female with Eisenmenger's syndrome, who had been treated with bosentan for 6 months. This treatment was stopped due to elevation of hepatic transaminases and when they returned to normal, sitaxentan was initiated. After four months of sitaxentan therapy, liver enzymes increased again and the ERA was stopped, but the enzymes continued to increase and the patient developed hyperbilirubinemia. She was started on corticosteroids, and made a rapid and sustained improvement. Subsequently, two other similar cases were published. In these two patients, liver biopsies showed extensive hepatocellular damage with eosinophil and lymphocyte infiltration, which was compatible with drug toxicity 32 Physicians should be aware that severe hepatic toxicity may be seen in patients receiving any of the current ERA therapies, and that ERA may interact with other potentially hepatotoxic medications. Monthly liver function tests are mandatory and patients must be advised to consult their prescribing physician urgently at the first signs and symptoms of hepatic toxicity, irrespective of previous normal liver function tests, even within a month earlier.34–36
As mentioned earlier, one of the most important drug interactions of sitaxentan is with oral anticoagulants. sitaxentan increases warfarin's half-life, with a consequent increase in international normalized ratio (INR). A recent analysis of data from STRIDE-2 and STRIDE-2X showed similar rates of bleeding events in PAH patients treated with either sitaxentan or bosentan, 5 but no difference in bleeding rate in patients that were receiving oral anticoagulation and those who were not. 37 Pulido et al 38 reported on the follow-up of 50 PAH patients that received sitaxentan and acenocoumarol (the most frequent oral anticoagulant used in various centers in Europe and Latin America). Following treatment, an INR > 5 in at least one INR determination was observed in 13 patients, although none of these patients had a clinically significant bleeding event. Two patients died of massive hemoptysis, but these episodes were not attributed to a drug interaction: both had Eisenmenger's physiology (which carries intrinsic bleeding abormalities39,40) and INR was kept constant at <2.0. These results suggest that coadministration of sitaxentan and acenocoumarol is clinically manageable and well tolerated.
Teratogenicity appears to be also a class effect of ERAs. In animal studies, disruption of endothelin receptor A or B isoforms during embryogenesis causes severe developmental abnormalities associated with an increment in perinatal morbidity.14,41 Experimental studies have not shown a carcinogenic effect in animals treated with sitaxentan. 41 Pregnancy should be excluded before initiation of treatment and prevented thereafter by use at least two methods of contraception. 15
In summary, sitaxentan showed a similar safety profile and tolerability to other ERAs, with a smaller increase in liver transaminases (average of 2%–4% for the 100 mg dose) and a manageable effect on INR.
Conclusions
PAH is still an incurable disease. However, new drugs have shown an improvement in patient survival. Sitaxentan is a selective ETA receptor antagonist that produces pulmonary vasodilatation, and inhibits vascular growth, remodeling and fibrosis. It has been shown to improve exercise capacity, functional class and hemodynamic parameters with a good safety profile. Also, long-term data at one and two years suggests that this improvement is maintained making it a good option for the treatment of patients with PAH. Future studies with sitaxsentan will focus on patients with PAH associated with collagen vascular disease and will evaluate combined sitaxentan and sildenafil therapy, with the intention to make it available to underserved communities suffering from PAH.
Disclosures
This manuscript has been read and approved by all authors. This paper is unique and not under consideration by any other publication and has not been published elsewhere. Dr. Pulido was a principal investigator of the STRIDE-4 study, is a consultant for Pfizer and has received research grants from Encysive (now owned by Pfizer) and Pfizer. Dr. Sandoval was a clinical advisor for Encysive (now part of Pfizer). The rest of the authors report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.
Footnotes
List of Abbreviations
6MWD, six-minute walk distance test; BID, twice a day; CPET, cardiopulmonary exercise testing; ERA, endothein receptor antagonists; ET, endothelin; ETA, endothelin A receptor; ETB, endothelin B receptor; INR, international normalized ratio; mPAP, mean pulmonary arterial pressure; OD, once a day; PAH, pulmonary arterial hypertension; PVR, pulmonary vascular resistance; PVRi, pulmonary vascular resistance, indexed; RAP, right atrial pressure; STRIDE, sitaxentan to relieve impaired exercise; ULN, upper limit of normal; VE, minute ventilation; VO2, oxygen consumption; WHO FC, World Health Organization functional class classification.