
Abstract
Raltegravir is the first integrase strand transfer inhibitor to be approved for the treatment of HIV infection. Administered orally in doses of 400 mg twice daily, it is well-tolerated and has minimal drug-drug interactions with coadministered antiretrovirals and other agents. In clinical trials including treatment-experienced and treatment-naïve HIV-infected adults, raltegravir in combination with other antiretroviral agents has demonstrated a rapid and potent virologic effect and a generally benign safety profile. Like other antiretrovirals, raltegravir should ideally be given with two additional agents to which the patient's virus is susceptible based on results of resistance testing. In this context, raltegravir offers a safe and effective option as a component of combination therapy in treatment-experienced patients who are infected with HIV-1 strains showing evidence of resistance to other antiretroviral agents. Pending the availability of longer-term efficacy and safety data, raltegravir cannot currently be recommended as part of first-line therapy for treatment-naïve patients.
Keywords
Introduction to Raltegravir
The current goal of antiretroviral therapy for both treatment-naïve and -experienced HIV-infected patients is achieving and sustaining an undetectable plasma HIV RNA level or viral load, i.e. below 40 or 50 copies/mL depending on the assay used.1,2 Achieving this therapeutic target has been shown to result in improvements in immune function as measured by increased CD4 T-cell counts, improved quality of life due to decreased incidence of AIDS-related morbidity, and increased survival. Recent improvements in the long-term safety and tolerability of available antiretroviral drugs make this a much more feasible goal. Even for heavily treatment-experienced patients who may harbor multidrug resistant HIV, designing effective combination regimens has become a possibility with newer generation drugs in the protease inhibitor (PI) and non-nucleoside reverse transcriptase inhibitor (NNRTI) classes (e.g. tipranavir, darunavir, etravirine) and agents from novel classes which are not cross-resistant with the original drug classes (e.g. enfuvirtide, maraviroc).3–10 However issues such as pill burden, tolerability, toxicity, and drug interactions can present a challenge when designing a regimen with 3 active drugs in the setting of multidrug resistant HIV.11,12 Newer antiretroviral agents that combine potency and efficacy with better safety and tolerability profiles continue to be required to optimize the long-term management of HIV disease.
Raltegravir (MK-0518, Isentress®) is the first inhibitor of a novel target, the HIV integrase enzyme, to be approved in many countries for use in combination with other antiretroviral agents for the treatment of HIV-1 infection in treatment-experienced adults who have evidence of viral replication and have HIV-1 strains resistant to multiple antiretroviral agents. 13
Review of Mode of Action, Pharmacology and Pharmacokinetics of Raltegravir
The HIV integrase enzyme catalyzes integration of viral DNA into host chromosomal DNA, a necessary process in replication of HIV-1 and HIV-2. 14 Integration occurs in several steps:14–16
Integrase binds to viral DNA following reverse transcription to form a pre-integration complex (PIC) with viral and cellular proteins
Integrase catalytically processes the 3‘-ends of viral DNA by cleaving two nucleotides from each 3’ end
The PIC enters the host cell nucleus
Integrase catalyzes the joining of 3’ end of viral DNA to the 5’ end of cellular DNA, a process called strand transfer
Unpaired nucleotides at 5’ ends of viral DNA are excised and single-strand gaps are repaired to form intact double-stranded DNA.
The integrase strand transfer inhibitors (InSTIs) raltegravir and elvitegravir (which is currently investigational) bind in the central catalytic domain of HIV integrase and specifically block the strand transfer step, resulting in circular DNA complexes (long terminal repeats or LTR circles) that are unable to bind to host DNA, and thereby irreversibly blocks viral replication. Consistent with its mechanism of action, raltegravir has been shown to produce a significant decrease in proviral DNA, unlike other antiretroviral agents 17 and also has activity against HIV-2.18,19
Because of its novel target, raltegravir retains activity against HIV that has reduced susceptibility to other available classes of antiretroviral agents, i.e. nucleoside reverse transcriptase inhibitors (NRTIs), NNRTIs, PIs, and entry inhibitors. Mutations within the active site of the integrase enzyme may confer resistance to raltegravir as well as to other integrase inhibitors including elvitegravir.20–23 However, as elvitegravir is currently only available through clinical trials and transmission of raltegravir-resistant HIV has not been reported to date, at the present time HIV with reduced susceptibility to raltegravir is not likely to be a concern in patients who have not previously received this agent.
Raltegravir is rapidly absorbed following oral administration, with a time to peak plasma concentration (Tmax) of approximately 3 hours in the fasted state. While a high-fat meal slows the rate of absorption resulting in a 7.4 hour delay in Tmax, it also extends the duration of absorption resulting in similar overall drug exposure as in the fasted state. Phase II and III studies were conducted without regard to food, consistent with the manufacturer's dosing recommendations. 13 Raltegravir pharmacokinetics have been shown to be similar regardless of gender, race, age (in adults and adolescents), HIV infection status, hepatic and renal function, and body mass index; however, these analyses were conducted in small numbers of patients.24–26
Raltegravir pharmacokinetics are appropriate for twice-daily dosing, with a terminal half-life of 7-12 hours and a slight degree of accumulation in 12 hour-concentrations (C12 h) after multiple doses.24,25 While the approved raltegravir dose is 400 mg twice daily, doses up to 800 mg twice daily were well-tolerated in Phase I studies. Considerable intra- and inter-subject variability in C12 h were observed in Phase III studies (coefficients of variation 122% and 212%, respectively), but interestingly, antiviral activity does not appear to correlate directly with raltegravir drug levels, 25 possibly due to the drug's unique mechanism of action as compared to other available antiretroviral agents.
Raltegravir is metabolized primarily by glucuronidation, mediated by the enzyme UGT1A1. 27 Modest, clinically insignificant increases in raltegravir plasma concentrations have been observed in individuals with decreased UGT1A1 activity (UGT1A1*28/*28 genotype) as compared to those with normal UGT1A1 activity (UGT1A1*1/*1). 28 Drugs that inhibit UGT1A1, including atazanavir, can increase raltegravir plasma levels, but in the case of atazanavir (with or without ritonavir boosting) this effect does not appear to be clinically relevant.29,30 Inducers of UGT1A1 can reduce raltegravir concentrations: coadministration of raltegravir and rifampin cannot currently be recommended due to an observed 40% reduction in the area under the plasma concentration curve (AUC) of raltegravir,2,13 an effect not compensated for by doubling the raltegravir dose, 31 and little clinical experience with their coadministration. Omeprazole coadministration increased raltegravir AUC by 210%; 32 however, no dose adjustment of raltegravir is recommended. 13 The UGT1A1 inducing effect of tipranavir/ritonavir results in lower raltegravir trough levels but does not appear to impact antiviral efficacy.33–35 Likewise the UGT1A1 inducers ritonavir and efavirenz have minimal impact on raltegravir pharmacokinetics and do not necessitate dose adjustments. 36 The PI lopinavir/ritonavir 37 and the CCR5 receptor antagonist maraviroc 38 have little impact on raltegravir levels in healthy volunteers. The NNRTI etravirine also had minimal effect on raltegravir levels in both healthy volunteers 39 and treatment-experienced HIV-infected patients. 34 The PI darunavir/ritonavir decreased raltegravir exposure by 29% but increased trough levels by 38% in six HIV-uninfected volunteers; 40 raltegravir trough levels were likewise not significantly decreased in the presence of darunavir/ritonavir among treatment-experienced HIV-infected subjects in two large studies (n = 83 and 125).34,35
Among NRTIs, tenofovir increases raltegravir exposure both in healthy volunteers 41 and HIV-infected treatment-naïve subjects (coadministered with lamivudine). 42 However, dose adjustment is not felt to be necessary. 41
Formal pharmacokinetic studies have not been conducted to evaluate the interactions between raltegravir and other antiretroviral agents, but no clinically significant interactions are expected with agents that are neither potent inducers nor inhibitors of UGT1A1. Raltegravir does not inhibit UGT1A1 enzymes so would not be expected to affect levels of coadministered drugs that are also metabolized by this system. 13 Unlike many other antiretroviral agents, raltegravir is not metabolized by nor does it induce cytochrome P450 (CYP) enzymes including CYP 3A4, nor does it inhibit P-glycoprotein-mediated transport.13,43 Therefore, drug interactions are not expected between raltegravir and drugs metabolized by or affecting these pathways.
In summary, the drug interactions with raltegravir that have been studied to date tend to be minimal and clinically insignificant, and do not necessitate dose adjustments for either raltegravir or the coadministered agent. The only drug where caution is advised when used with raltegravir is rifampin. 13
No dose adjustment appears to be necessary in patients with moderate hepatic insufficiency or severe renal insufficiency, based on available pharmacokinetic data. 13
Efficacy Studies
Treatment naïve
The first clinical study to examine the effect of raltegravir in treatment naïve adults was Protocol 004.42,44 The study included subjects with plasma HIV-1 RNA ≥ 5000 copies/mL, CD4 counts ≥100 cells/mm3, and no more than 7 days of previous antiretroviral therapy. Randomization was stratified for baseline HIV RNA levels ≤ or >50,000 copies/mL. In the first part of this multicentre, double-blind, randomized, placebo-controlled, dose-finding study, 35 subjects were randomized to either placebo or raltegravir 100 mg, 200 mg, 400 mg, or 600 mg twice daily as monotherapy for 10 days (n = 6-8 in each group). Mean HIV RNA at baseline was 4.53 to 4.97 log10 copies/mL and mean baseline CD4 counts were 256 to 569 cells/mm3 in all groups. After 10 days of monotherapy, the combined raltegravir dose groups (n = 28) had mean viral load reductions of 2.0 log10 copies/mL from baseline, significantly greater than that observed with placebo (n = 7) (p < 0.001 for the comparison with each raltegravir dose arm). 44 HIV RNA levels <400 copies/mL were reached at day 10 by 50%-57% of subjects randomized to raltegravir, as compared to none of those randomized to placebo. At least one subject in each raltegravir dose group, and none in the placebo group, had viral load < 50 copies/mL by day 10. Raltegravir trough levels in plasma were dose-dependent, increasing from the 100 mg to the 600 mg dose, and drug exposure (AUC) increased with dose from 100 mg to 400 mg (400 mg = 600 mg); however, viral load reductions were not different among the different raltegravir dose groups. The slope of the viral decay curve over 10 days was also similar in all raltegravir treatment groups and greater than in the placebo arm, and did not differ according to entry HIV RNA level < or >50,000 copies/mL.
The 004 study was continued into a second part, wherein 30 subjects from Part I either continued on the same dose of raltegravir with the addition of tenofovir 300 mg and lamivudine 300 mg once daily, or if randomized to placebo in Part I, received efavirenz with tenofovir and lamivudine (Table 1). Part II of the study included an additional 171 treatment-naïve subjects who were randomized to receive either one of the same 4 raltegravir doses as in Part I or efavirenz 600 mg once daily, in combination with tenofovir and lamivudine. Entry criteria were the same as in Part I, with the addition of genotypic sensitivity to efavirenz, tenofovir, and lamivudine, and groups were again stratified for baseline viral load ≤ or > 50,000 copies/mL. After 48 weeks, all patients in the raltegravir groups received raltegravir 400 mg twice daily. 45
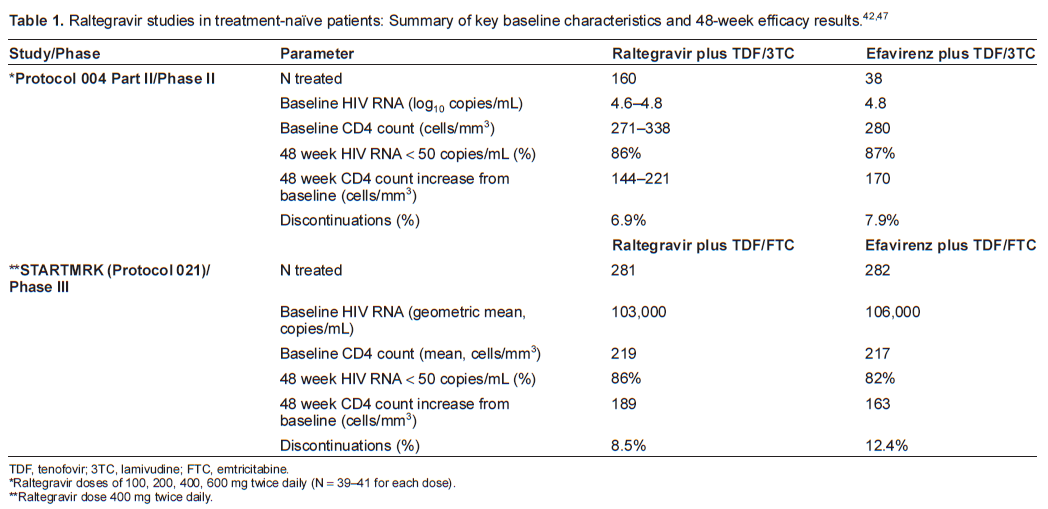
TDF, tenofovir; 3TC, lamivudine; FTC, emtricitabine.
Raltegravir doses of 100, 200, 400, 600 mg twice daily (N = 39-41 for each dose).
Raltegravir dose 400 mg twice daily.
After 48 weeks of combination therapy, proportions of subjects with HIV RNA < 400 copies/mL (the primary endpoint of the study) and <50 copies/mL were similar across all treatment arms (85%-98% and 83%-88%, respectively) (Table 1). 42 Of note, viral load reductions were more rapid with raltegravir than with efavirenz: subjects receiving any dose of raltegravir achieved HIV RNA < 50 copies/mL earlier than patients receiving efavirenz (p < 0.05). In addition, the proportion of subjects with viral load < 50 copies/mL was greater in each of the raltegravir groups than the efavirenz group at weeks 2, 4, and 8, 42 and plasma HIV RNA levels were lower for the combined raltegravir groups vs. the efavirenz group at all time points up to day 168. 46 By week 24, the raltegravir and efavirenz arms had similar levels of viral load reduction (85%-95% in the raltegravir groups and 92% in the efavirenz group had HIV RNA < 50 copies/mL). 42 These virologic responses were sustained to week 96 (83%-84%). 45 Results were similar regardless of HIV RNA at entry (≤ and > 50,000 copies/mL, and ≤ and > 100,000 copies/mL). 42 CD4 count increases were similar in all treatment groups: mean change from baseline to week 96 was 221-232 cells/mm3. 45
More recently, raltegravir 400 mg twice daily (the dose selected for Phase III studies) was compared with efavirenz 600 mg once daily, this time with a backbone of tenofovir and emtricitabine, among 563 treatment-naïve subjects in the double-blind, randomized STARTMRK study (Protocol 021) (Table 1). 47 Entry criteria included baseline HIV RNA > 5000 copies/mL and susceptibility to efavirenz, tenofovir, and emtricitabine. The raltegravir arm was shown to be non-inferior to the efavirenz arm in terms of the proportion of subjects with viral load < 50 copies/mL after 48 weeks of treatment (non-inferiority p < 0.001) (Table 1). Again, time to viral load < 50 copies/mL was shorter with raltegravir than with efavirenz (log-rank p < 0.001). 47 Raltegravir also resulted in greater CD4 cell count increases at week 48 than efavirenz (95% confidence interval [CI] for the difference between arms 4-47 cells/mm3). Discontinuation rates were lower in the raltegravir arm (8.5%) than in the efavirenz arm (12.4%) and fewer patients discontinued raltegravir for lack of efficacy (n = 4 vs. n = 2 for efavirenz).
The more rapid initial reduction in viral load with raltegravir as compared to efavirenz observed in the treatment-naïve studies suggests a unique effect on the dynamics of viral decay. 46 The mechanism of rapid viral load reduction with raltegravir remains to be elucidated, and the clinical significance in terms of longer-term outcomes is unknown, given that by week 24 the virologic response rates are similar with efavirenz- as with raltegravir-containing regimens.42,47
Treatment experienced HIV-infected adults
The safety and efficacy of raltegravir were studied among 178 treatment-experienced adults in a Phase II, randomized, double-blind, placebo-controlled, dose-ranging study, Protocol 005 (Table 2). 29 Subjects had plasma HIV RNA > 5000 copies/mL on their current stable (>3 months) antiretroviral therapy, CD4 cell count > 50/mm3, and virus demonstrating genotypic and phenotypic resistance to at least one drug from each of the NRTI, NNRTI, and PI classes. Eligible subjects were randomized to receive either placebo or one of three doses of raltegravir (200, 400, or 600 mg) twice daily. All patients received an optimized background regimen (OBR) selected by the investigator based on resistance test results. Randomization was stratified by presence or absence of enfuvirtide in the OBR and by resistance to 1 PI or more than 1 PI at baseline. During the 005 study, 400 mg twice daily was selected as the raltegravir dose to be used in Phase III trials; thereafter, patients who had received at least 24 weeks of treatment (with any dose of raltegravir or placebo) in the study could receive open label raltegravir 400 mg twice daily. 48 In the case of virologic failure after week 16, defined as viral load reduction <1.0 log10 copies/mL from baseline, subjects could switch to open label raltegravir plus OBR.

OBR, optimized background regimen.
Raltegravir doses of 200, 400, and 600 mg twice daily (N = 43-45 for each dose).
Raltegravir dose 400 mg twice daily.
Participants in study 005 were randomized into one of two substudies, one including atazanavir in the OBR and the other not including atazanavir. This was based on earlier pharmacokinetic data indicating that atazanavir increases raltegravir plasma concentrations, 30 the clinical significance of which was unknown at the time. When it was determined that difference in response rates between the raltegravir and placebo arms was not affected by the presence or absence of atazanavir in the OBR, the results of the two substudies were combined in a pooled analysis, as per the a priori plan. 29
At baseline, the 178 treatment-experienced patients included in the pooled analysis of study 005 had a mean plasma viral load of 50,000 copies/mL and a mean CD4 count of 240 cells/mm3, with no notable differences between treatment groups (Table 2). 29 The OBR included from 2 to 7 agents (median 4); however, 72% of treated subjects had no active agents in the OBR by genotypic testing and 48% based on phenotypic testing, both with the exception of enfuvirtide. At the end of the 24-week double-blind treatment period, mean decreases in viral load were 1.80 to 1.87 log10 copies/mL in the raltegravir dose groups and 0.35 log10 copies/mL in the placebo group, and the proportions of subjects with viral load < 50 copies/mL at week 24 were greater with each dose of raltegravir than with placebo (p < 0.0001 for all comparisons) (Table 2). The antiviral effect of raltegravir at all doses was observed as early as 2 to 4 weeks and was sustained through week 24, with no clear evidence of a dose-response relationship. CD4 cell counts increased from baseline to 24 weeks to a greater extent in the raltegravir groups than in the placebo group (p < 0.0001 for all comparisons). Treatment differences were observed consistently across all degrees of baseline HIV drug resistance, as assessed by phenotypic and genotypic sensitivity scores (PSS, GSS).
Results are available to 48 weeks for 178 patients who took open-label raltegravir 400 mg twice daily from weeks 24 to 48 following the 24-week double-blinded phase of the 005 study. 48 Patients who had virologic suppression at 24 weeks generally maintained it to 48 weeks. Overall 54% of patients who had been randomized to receive raltegravir during the first 24 weeks, and 9% of those randomized to receive placebo, had viral load < 50 copies/mL at week 48.
The Blocking Integrase in Treatment Experienced Patients with a Novel Compound against HIV, Merck (BENCHMRK) studies are two ongoing randomized, double-blind, placebo-controlled Phase III trials with a planned duration of 156 weeks (Table 2).49,50 BENCHMRK-1 is being conducted in Europe, Asia, Australia, and Peru, and BENCHMRK-2 in North and South America. These identical concurrent studies enrolled treatment-experienced patients at least 16 years of age with viral load greater than 1000 copies/mL and evidence of phenotypic or genotypic resistance to at least one agent in each of the NRTI, NNRTI, and PI classes. Eligible subjects were randomized in a ratio of 2:1 to receive either raltegravir 400 mg or placebo twice daily along with an OBR selected by the investigator based on previous antiretroviral treatment and results of resistance and other laboratory tests. The OBR could include darunavir or tipranavir, both of which were investigational at the time the studies enrolled. Randomization was stratified by use of enfuvirtide in the OBR (yes/no) and degree of resistance to PIs (resistance to 1 PI vs. >1 PI).
Results from the 699 subjects included in both BENCHMRK studies were pooled in a pre-specified analysis 49 (Table 2). Baseline characteristics were generally well-balanced between the raltegravir (n = 462) and placebo (n = 237) groups. Overall 88% of study subjects were male and the median age at study entry was 45 years. Study participants had received a median 10 years of previous antiretroviral therapy including a median of 12 drugs, and over 90% had a previous history of AIDS. Subjects received a median of 4 antiretroviral drugs in their OBR (range 1 to 7). Approximately 20% of patients in both arms received enfuvirtide in their background regimen and were previously naïve to this agent, and approximately 37% of patients received darunavir and were darunavir-naïve.
Consistent results were observed in BENCHMRK -1 and BENCHMRK-2. In the combined 48-week analysis, raltegravir plus OBR resulted in mean viral load reductions of 1.7 log10 copies/mL vs. 0.8 log10 copies/mL for placebo plus OBR (p < 0.001). 49 The proportions of patients with viral load < 50 copies/mL at 48 weeks was greater with raltegravir than with placebo (p < 0.001) (Table 2). 50 Mean CD4 increases from baseline to week 48 were also greater with raltegravir than with placebo (p < 0.001). 49
Raltegravir plus OBR produced better virologic suppression (<50 copies/mL) than OBR alone in almost all subgroups examined, including when results were analyzed by baseline viral loads (≤ vs. > 100,000 copies/mL or ≤ vs. > 50,000 copies/mL), baseline CD4 cell counts (≤50, >50 and ≤200, or >200 cells/mm3), number of active PIs in OBR (0 vs. 1 or more), PSS (0, 1, or 2), and GSS (0, 1, or 2). 50 In these analyses, enfuvirtide and darunavir were each counted as one active agent, and darunavir was counted as an active PI, when included in the OBR of subjects naïve to either drug. The only subgroups where raltegravir did not demonstrate a virologic benefit over placebo were those with PSS or GSS ≥ 3 in the background regimen. This result is not surprising and parallels the findings of trials of other antiretroviral agents that were new at the time they were studied (including enfuvirtide, tipranavir, etravirine, darunavir).3–9 The effect of the new agent is most pronounced in the setting of a relatively weak background regimen, one with no or only one active agent. Of particular note, even among patients in BENCHMRK with no active drugs in their background regimen (PSS = 0), 51% of those who received raltegravir had viral load < 50 copies/mL at 48 weeks, as compared to only 2% of placebo recipients. 49 Response rates at 48 weeks in the raltegravir arms of the BENCHMRK studies improved to 61% and 71% with increasing PSS to1 and ≥2, respectively, indicating the importance of having two active agents if possible in the background regimen to support raltegravir's antiviral effect. The results of BENCHMRK and other new drug studies illustrate that the incremental effect of adding raltegravir or any other new active drug is minimal in subjects receiving an effective OBR comprising 3 or more active drugs.
With regard to clinical endpoints in the BENCHMRK studies, there was a trend towards fewer AIDS-defining conditions in the raltegravir arms than in the placebo arms over 48 weeks: exposure-adjusted rates of AIDS-defining conditions (most commonly esophageal candidiasis) were 3.7 per 100 patient-years and 6.2 per 100 patient-years, respectively [relative risk (RR) = 0.6; 95% CI 0.3-1.4]. 51 The median time to onset of AIDS-defining conditions was 64 days in the raltegravir arms vs. 105 days in the placebo arms. Higher viral load and lower CD4 cell counts at baseline, and previous AIDS-defining conditions were seen more often in subjects who developed an AIDS-defining condition or who died. These patients also had poorer virologic and immunologic responses to study treatment.
Recent observational data support the efficacy of raltegravir in treatment-experienced patients. A prospective cohort of 99 3-class (NRTI, NNRTI, and PI) experienced patients showed that 65% (39/65) of subjects who received raltegravir in their new antiretroviral regimen achieved a viral load < 50 copies/mL after 24 weeks of treatment, while only 38% (15/39) of those who did not receive raltegravir achieved this level of suppression. 52 Half of the patients in this cohort received darunavir and a third received another PI. Inclusion of raltegravir in the regimen was associated with a greater likelihood (odds ratio 3.1, 95% CI 1.1, 8.6) of virologic suppression by multivariate logistic regression. CD4 increases at 24 weeks were also greater with raltegravir-containing regimens (mean 71 ± 135 cells/mm3) than with those not containing raltegravir (mean 39 ± 126 cells/mm3). Similarly good responses were observed in a multicenter, retrospective analysis of 53 triple-class resistant patients receiving raltegravir and etravirine together through their respective expanded access programs. 53 A boosted PI was included in the regimen of 89% of patients (47/53), mainly darunavir (44/53, 83%), and enfuvirtide was included in 11% (6/53). By intent to treat analysis at week 24, 94% (50/53) of patients had viral load < 50 copies/mL and the mean CD4 increase was 86 cells/mm3. Similar results were observed among 103 triple class resistant patients treated with a regimen including raltegravir, etravirine, and darunavir/ritonavir in the single arm TRIO study. 54 Some subjects also received one or more NRTI (73%), enfuvirtide (4%), or both (10%). After 24 weeks of treatment, 90% (93/103) of these treatment experienced subjects achieved viral load loads < 50 copies/mL, and the median CD4 cell increase was 99 cells/mm3 (IQR 32, 147).
The antiviral efficacy of raltegravir can be adversely impacted by viral mutations that lead to changes in the integrase enzyme, possibly affecting the structure of the enzyme's active site or its interactions with integrase inhibitors. 55 Treatment-emergent mutations observed in clinical trials among patients experiencing virologic failure on a raltegravir-based regimen occurred along mutational pathways defined by one of N155H, Q148H/R/K, or Y143C/H/R along with one or more additional mutations.21,23,50,56,57 In virus resistant to raltegravir from patients failing therapy in the BENCHMRK studies, single mutations were rare, and two or more integrase mutations were usually identified; no single mutation had a significant impact on resistance or virologic response. 50 Each mutation at Y143, Q148, and N155 has a severe impact on viral replicative capacity; single mutants therefore are unfit and present in numbers too small to be detected by population-based genotypic assays. The secondary mutations in each pathway increase viral replicative fitness as well as further reducing susceptibility to raltegravir.
Thus high-level resistance to raltegravir requires a mutation at position N155, Q148, or Y143 and at least one additional mutation. The most common mutational pattern observed among raltegravir treatment failures in study 005 was Q148H/G140S, which resulted in a loss of susceptibility to raltegravir of greater than 100-fold. 21 The highest levels of resistance to raltegravir in the 005 study were observed in the presence of three or more mutations.
Among previously antiretroviral naïve subjects who then failed treatment including raltegravir, only 2 subjects in the 004 study developed raltegravir-associated mutations, both having N155H. 42 No new raltegravir-associated mutations emerged in this study between 48 and 96 weeks. 45 In the STARTMRK study, 12 previously treatment naïve subjects failed raltegravir-based therapy, of whom 4 developed mutations conferring resistance to raltegravir. 47 The mutational patterns were similar to those observed among treatment-experienced patients.
Cross-resistance between raltegravir and the investigational integrase inhibitor, elvitegravir, has been identified in analysis of isolates from treatment-experienced patients and clonal analysis.20,22,23 This cross-resistance between integrase inhibitors appears to be clinically relevant: two patients who failed an elvitegravir-containing regimen subsequently had no virologic response to raltegravir. 58 Mutations at positions 148 and 155 confer resistance to both raltegravir and elvitegravir, and certain double mutants (G140S/Q148H, G140S/Q148R) are highly resistant to both agents. 21 Research is underway to develop a new generation of integrase inhibitors with activity against elvitegravir- and raltegravir-resistant virus,23,59 including integrase inhibitors with different mechanisms of action, such as blocking the formation of the pre-integration complex. 60
Safety and Tolerability
A single supratherapeutic dose of raltegravir (1600 mg) does not prolong the QTc interval in healthy volunteers. 61
Raltegravir in doses of 100 to 600 mg twice daily as monotherapy was well-tolerated in 28 previously treatment-naïve subjects in the 004 study. 44 Dizziness (n = 3), fatigue and headache (n = 2 each) were the most common adverse events among patients taking raltegravir. Most adverse events were mild to moderate and none were dose-related. There were no serious adverse events or discontinuations related to intolerance or toxicity during the 10-day monotherapy phase of this study. In the 48-week extension phase, no dose-related toxicities were observed when raltegravir was administered in combination with tenofovir and lamivudine. 42
Available clinical trial data from the 004 and STARTMRK studies allow a comparison of the safety profile of raltegravir with that of efavirenz when either is given as part of combination therapy (with tenofovir plus either lamivudine or emtricitabine) to treatment-naïve subjects.42,45,47 Overall, raltegravir was better tolerated than efavirenz, with fewer drug-related clinical events, particularly fewer neuropsychiatric adverse experiences (including dizziness, headache, abnormal dreams, and nightmares) and less frequent rashes. Laboratory abnormalities generally occurred at comparably low rates with raltegravir and efavirenz.42,45,47 After 96 weeks in the 004 study, elevated pancreatic amylase >2 × the upper limit of normal (ULN) and lipase >3 × ULN were seen in 4 and 3, respectively, of the 160 patients receiving raltegravir (2.5% and 1.3%, respectively), while neither was observed in any patient who received efavirenz. 45 Increases in creatinine kinase to at least 10 × ULN were also seen more frequently with raltegravir (10/160, 6.3%) than with efavirenz (1/38, 2.6%). Drug-related increases in alanine transaminase (ALT) to >5 × ULN were observed more frequently in the raltegravir 200 mg dose group during the first 24 weeks of the 004 study (10.0% as compared to 5.3% in the efavirenz group). 42 However, ALT increases were observed at rates of 0, 0 and 5.0% in the 100 mg, 400 mg, and 600 mg raltegravir dose groups, respectively, and ALT abnormalities were less common with raltegravir than with efavirenz at 96 weeks (1.3% vs. 5.3%). 45 In the STARTMRK study, severe elevations in ALT and aspartate aminotransferase (AST) occurred infrequently (1.8% and 2.1%, respectively) with raltegravir 400 mg twice daily. 47
These studies among treatment-naïve subjects permit the best assessment of the effects of raltegravir on serum lipids, unaffected by previous antiretroviral therapy. Increases in total cholesterol, LDL cholesterol, and triglycerides were greater in the efavirenz arms than in the raltegravir arms of both the 004 and STARTMRK studies during the first 48 weeks; however, greater increases in HDL cholesterol were observed with efavirenz, and the mean decrease in total to HDL cholesterol ratio was similar with efavirenz and raltegravir.42,47 Similar comparisons between raltegravir and efavirenz were observed after 96 weeks in the 004 study, although the difference in the impact on triglycerides (mean decrease with raltegravir and mean increase with efavirenz) was no longer statistically significant. 45 Overall, the impact of 48 to 96 weeks of raltegravir on serum lipids is minimal and probably not clinically relevant.42,45,47
Safety data from clinical trials in treatment-experienced HIV-infected subjects include 507 patients who received raltegravir 400 mg twice daily and 282 who received placebo as comparators, in Protocol 004 and the BENCHMRK studies. Overall raltegravir was well-tolerated with a safety profile similar to that of placebo, and most drug-related events were mild to moderate in severity.29,49 Adverse events led to treatment discontinuation in 2.0% of treatment-experienced subjects who received raltegravir and 1.4% of subjects who received placebo. 13
Severe (DAIDS severity Grade 3 or 4) laboratory abnormalities, including liver enzyme elevations, were observed infrequently in treatment-experienced subjects randomized to receive raltegravir in these clinical trials.29,49 Grade 3/4 AST and ALT abnormalities (elevations to >5 × ULN) were seen at rates of 3%-4% in patients randomized to either raltegravir or placebo in the combined BENCHMRK studies. 49 Not surprisingly, AST and ALT elevations during Phase III studies were more common in both raltegravir- and placebo-treated subjects coinfected with hepatitis B and/or C. 13
Grade 2 or greater increases in creatine kinase (≥6 × ULN) as well as myopathy and rhabdomyolysis have been observed in raltegravir-treated subjects in clinical trials, 13 and a case report has been published of rhabdomyolysis possibly associated with raltegravir. 62 Across studies in treatment-experienced subjects, Grade 4 elevations in creatine kinase (≥20 × ULN) were seen more frequently with raltegravir (2.2%) than with placebo (0.7%). 13 The contribution of raltegravir to creatinine kinase elevations and symptomatic muscle disease is difficult to evaluate in patients receiving multiple antiretrovirals and other drugs concomitantly. The manufacturer recommends caution when prescribing raltegravir to patients at risk for muscle disease. 13
In the 16-week analysis of the BENCHMRK studies and a subsequent analysis presented to the United States Food and Drug Administration (FDA) of all patients exposed to raltegravir in the double-blind portion of Phase II and III studies, an imbalance was noted in the occurrence of malignancies between raltegravir and the comparator arms. 63 Among 758 patients receiving raltegravir representing 508 person-years of exposure, 10 malignancies were observed, equalling 2.0 cases per 100 person-years. Only one malignancy occurred among the 323 patients with 169 person-years on the comparator, representing an incidence rate of 0.6 per 100 person-years. The relative risk of malignancy with raltegravir vs. comparator was observed to be 3.3 (95% CI 0.5, 144). However, 9 of the 10 cases occurred within the first 3 months of raltegravir therapy, suggesting a pre-existing condition, and 3 cases were recurrences. Also there was no specific pattern to the diagnoses, which included malignancies expected in the setting of advanced immunodeficiency such as Kaposi's sarcoma, lymphoma, and squamous carcinomas. The association was no longer found in a later analysis of the same patients with longer follow-up: a total of 19 cases during 820 person-years with raltegravir (2.2/100 patient-years) and 5 cases during 261 person-years on comparator (1.8/100 patient-years) were observed, giving a relative risk of 1.2 (95% CI 0.4, 4.1). 49 In the combined BENCHMRK study analysis, malignancies were diagnosed in 3.5% (16/462) of treatment experienced subjects who received raltegravir and 1.7% (4/237) of those who received placebo. 49 The relative risk of cancer with raltegravir vs. placebo was calculated to be 1.54 (95% CI 0.50, 6.34). Again the cancers seen in the raltegravir arms were mainly squamous cell (n = 9), lymphoma (n = 3), and Kaposi's (n = 2). In 14/16 cases of malignancy in the raltegravir arms, by the time of diagnosis the patients had shown a brisk virologic response to antiretroviral therapy and a CD4 increase of at least 50 cells/mm3, leading to the theory that immune reconstitution may have been a contributing factor. In treatment-naïve subjects, the malignancy rate after 96 weeks in the 004 study was lower among subjects treated with raltegravir (3/160 = 1.9%) than among those who were treated with efavirenz (1/38 = 2.6%), 45 and in the STARTMRK study, only one malignancy was seen in the first 48 weeks in the 281 subjects treated with raltegravir (0.4%) as compared to 9 in the 282 treated with efavirenz (3.2%). 47 On balance with the currently available evidence, the association of raltegravir with malignancy does not appear to be strong, and is likely mediated by other factors including immune reconstitution. However further research over longer durations of therapy is required to elucidate whether such an association exists.
With regard to safety of raltegravir in pregnancy and breast-feeding, no controlled data are available. In animal studies using exposures 3-4 times greater than those at the recommended human dose, the drug did not have teratogenic or post-partum effects other than treatment-related increases in supernumerary ribs in rats. 13 The FDA has assigned raltegravir to Pregnancy Category C, indicating that animal studies have revealed an adverse effect on the fetus, but benefits in humans may be acceptable despite potential risks. The safety of raltegravir in breast-feeding is likewise unknown. It is secreted in the milk of lactating rats, resulting in drug concentrations in milk of approximately 3-fold higher than in plasma. 13
Patient Focussed Perspectives
The first antiretroviral agent to become available from outside the three original classes (NRTI, NNRTI, and PI) was the fusion inhibitor enfuvirtide. Representing a novel class without cross-resistance to the other three classes, this drug was highly beneficial as an active component of a salvage regimen in some instances of multidrug resistant HIV.3,4 However its acceptability was hampered by the need for twice daily subcutaneous injections and the attendant needle-related problems, including bothersome and often persistent injection site reactions.64,65 When raltegravir became available, it presented an attractive alternative to enfuvirtide, again belonging to a new class without cross-resistance to the earlier classes, and with the added advantages of oral administration and generally good tolerability in clinical trials. More than 100 treatment-experienced patients on successful enfuvirtide-based salvage regimens have been able to replace the enfuvirtide component of their regimen with raltegravir.66–68 This strategy of replacing an injectable agent with an oral one has generally been successful in maintaining virologic suppression for 6 months or more, as well being highly tolerable and acceptable from the patient's point of view. However, in one case series, three cases of hepatotoxicity were observed within 2-4 weeks of switching from enfuvirtide to raltegravir, possibly due to increased plasma levels of co-administered tipranavir/ritonavir. 69 On balance, the strategy of replacing enfuvirtide with raltegravir within a virologically successful regimen (with the possible exception of regimens including tipranavir/ritonavir) appears to be safe and effective.
Neuropsychiatric symptoms, including depression and suicidal ideation, were observed in previously antiretroviral-naïve patients treated with raltegravir in the 004 and the STARTMRK studies, at rates of 8% and 10%, respectively, during the first 8 weeks of treatment.42,47 With longer follow-up in the 004 study, the rates of neuropsychiatric adverse events increased to 13% at 48 weeks and 16% at 96 weeks.42,45 The occurrence of these symptoms was less frequent with raltegravir than in the efavirenz comparator arms, but given that such symptoms are known to occur at a high frequency during the first few weeks of efavirenz therapy, 70 the rates with raltegravir should also be considered clinically relevant. Unfortunately the clinical trial reports present neuropsychiatric symptoms as a group and do not give specific rates for depression-related events (e.g. major depression, suicidal ideation, attempted or completed suicide) so it is not possible to tease out the incidence of these events with raltegravir in clinical trials.42,45,47 On the other hand, in the 005 and BENCHMRK studies among treatment-experienced patients, depression was seen at a similarly low frequency with raltegravir (2.6%) as with placebo (2.8%). 13 A recent report describes exacerbation of pre-existing depression temporally related to starting raltegravir in four HIV-infected men, one of whom was hospitalized for suicidal risk. 71 Before starting raltegravir all four patients were being treated with a number of psychoactive medications, and in each case the depression resolved with continued raltegravir therapy. Whether raltegravir may have precipitated a relapse in depression by some hereto undescribed drug interaction or some other mechanism is unknown. However, given the high prevalence of depression in the HIV-infected population, vigilance for a relapse in depressive symptoms may be warranted, particularly during the first few weeks of therapy with raltegravir.71,72
Conclusions and Place in Therapy
Raltegravir is currently approved for use in combination with other antiretroviral agents for the treatment of HIV-1 infection in treatment-experienced adults who have evidence of viral replication and have HIV-1 strains resistant to multiple antiretroviral agents. 13 This recommendation is supported by the data from Phase II (005) and Phase III (BENCHMRK) studies conducted in patients with triple-class resistant HIV who were failing their current antiretroviral regimen, demonstrating the superior efficacy of raltegravir as compared to placebo in suppressing HIV viral load when either is combined with an individually selected background antiviral regimen. In the BENCHMRK studies, the superiority of the virologic response to raltegravir over that of placebo was not affected by baseline viral load or CD4 cell count. 50 Optimal efficacy of raltegravir was observed when it was given with a background regimen containing 2 (vs. 0 or 1) active drugs, but further meaningful improvements were not observed with 3 or more concomitant active agents. This finding is not exclusive to raltegravir, and supports the recommendation in current guidelines that antiretroviral therapy regimens should comprise 3 active agents if at all possible.1,2 Thus clinical trial data support the use of raltegravir, ideally in combination with 2 other active agents, in treatment-experienced patients with 3-class resistant virus. Data from clinical trials and observational studies, as well as available pharmacokinetic data, support the effective use of most available antiretrovirals (including enfuvirtide, darunavir/ritonavir, etravirine, and maraviroc) in combination with raltegravir, without additional safety concerns or dose adjustment. The possible exception is tipranavir/ritonavir which may lead to increased hepatotoxicity in combination with raltegravir, although some have used this combination successfully. Increased vigilance for hepatic adverse events may be advisable if this combination is selected.
The safety profile of raltegravir is remarkably benign in both treatment-naïve and -experienced patients, with up to 96 weeks of data. In treatment-naïve patients, raltegravir caused less clinical adverse events, particularly neuropsychiatric symptoms, than efavirenz, and similarly low rates of laboratory abnormalities. The only exceptions were more frequent abnormalities of pancreatic lipase, amylase and creatine kinase with raltegravir. However, raltegravir was not associated with elevated pancreatic enzymes or clinical pancreatitis in the treatment-experienced trials. Elevated creatine kinase was observed more frequently with raltegravir than placebo in treatment-experienced studies, and clinical myopathy and even rhabdomyolysis have been observed in association with this agent, although apparently rarely. Hepatic enzyme abnormalities occurred at low rates with raltegravir in clinical trials, similar to efavirenz and placebo in the naïve and experienced studies, respectively. Unlike many current antiretroviral agents (including most PIs, efavirenz, and stavudine), raltegravir has a minimal and clinically benign effect on lipid profiles. An early association of raltegravir with malignancy in treatment-experienced patients has not been borne out by longer-term analysis, and in treatment-naïve studies malignancy rates were lower in the raltegravir than in the efavirenz arms. A possible association of raltegravir with depression is suggested by results of treatment-naïve studies and case reports in treatment experienced patients, but remains to be verified.
Although direct evidence from clinical trials is not available, the favorable results particularly with regard to safety could logically be extended to recommend the use of raltegravir in treatment-experienced patients who are not triple class resistant, but who require it to form a regimen with 3 active agents when other alternatives are not an option due to established intolerance or toxicity issues. Similarly, in treatment-experienced patients on a virologically successful regimen who require substitution of one active agent with another for reasons of ongoing intolerance or toxicity associated with a single agent in the regimen, raltegravir could be considered an appropriate replacement. Some observational data support its use as a replacement for enfuvirtide in this setting, and results of controlled clinical trials testing this strategy are pending.
With regard to its use in treatment-naïve patients, clinical trial data are available demonstrating the efficacy of raltegravir in combination with 2 nucleosides (specifically tenofovir plus lamivudine or emtricitabine) as first-line therapy.42,45,47 The efficacy of raltegravir-based regimens in this context is comparable to that of the gold standard efavirenz-based regimens that are recommended in current guidelines.1,2,73 The excellent tolerability of raltegravir demonstrated in clinical trials would be a benefit in first-line therapy, as it would be likely to support adherence and ultimately better long-term outcomes. However the longest data available for use of raltegravir in treatment-naïve patients is 2 years. 45 In the context of first-line therapy, other considerations are also important, particularly given the number of other currently available treatment options that are supported by clinical experience and long-term data. For example, convenience is an important consideration from the patient's perspective, and enhanced adherence with simpler regimens has been shown to result in better long-term outcomes. With the availability of efficacious once daily regimens, the twice daily dosing requirements of raltegravir put it at a disadvantage as a first-line option. However, the potential for once daily dosing is supported by data indicating that once raltegravir is bound to integrase, it takes a long time to dissociate, suggesting that its antiviral effect may be prolonged. 74 An ongoing clinical trial comparing once daily to twice daily raltegravir in combination with 2 nucleosides for treatment naïve patients may help to resolve this issue (ClinicalTrials.gov number NCT00745823).
Perhaps even more importantly, when selecting a regimen for a treatment-naïve patient, safety becomes a critical factor. Given that patients who commence antiretroviral therapy today are likely to continue taking it for many years, demonstrated long-term safety is particularly crucial. With this in mind, relatively new agents such as raltegravir are at a disadvantage as compared to established agents for which safety data are available for patients receiving these regimens over many years. While clinical trial data for raltegravir covering up to 2 years of treatment are reassuring, the potential effects of longer term exposure are presently unknown. Given that raltegravir is the first available agent in the integrase inhibitor class, its safety profile cannot be supported by longer-term data from other similar agents. The validity of safety concerns related to its novel mechanism of action and effect on the host cell nucleus are currently unknown. In view of these concerns with regard to long-term safety and based on the data currently available, raltegravir cannot be recommended at this time as a component of first-line therapy for treatment-naïve patients.
In summary, raltegravir is the first integrase inhibitor to become available for the treatment of HIV. It is well-tolerated with oral administration and has minimal drug interactions. In combination with other antiretroviral agents, it has demonstrated a rapid and durable virologic suppression and a good safety profile over 96 weeks of treatment. Raltegravir offers a safe and effective option as a component of combination therapy in treatment-experienced patients, in whom it should be given with ideally 2 additional active agents. Before its use can be extended to first-line therapy for treatment-naïve patients, longer-term efficacy and safety data, including investigation of once daily dosing, will be required.
Disclosure
The author has received honoraria for speaking engagements and/or advisory boards from Abbott, Bristol-Myers Squibb, Boehringer-Ingelheim, Gilead, GlaxoSmith Kline, Merck, Pfizer, and Tibotec.