
Abstract
Raltegravir is the first antiretroviral drug in the class of integrase inhibitors approved for the treatment of human immunodeficiency virus type 1 (HIV-1) in combination with other antiretroviral agents in treatment-experienced adults with evidence of ongoing viral replication and resistance to multiple antiretroviral drugs. Since raltegravir has a different mechanism of action to the currently licensed antiretroviral agents, it is a welcome addition in the treatment of HIV-1. Results from clinical studies to date indicate that raltegravir exhibits potent antiviral activity particularly against HIV-1 strains which exhibit resistance to other classes of antiretroviral drugs. It is well tolerated and has a favorable safety profile. Long-term follow-up data on its resistance profile and on potential interactions with other antiretroviral as well as concomitant medications will ultimately define its future role in the treatment of HIV-1 infection. This review briefly describes the mechanism of action of raltegravir and its pharmacokinetic profile, summarizes efficacy and safety data from recent clinical trials and implications for the use in treatment-naïve as well as treatment-experienced patients, depicts raltegravir's emerging resistance profile, and highlights potential drug-drug interactions.
Introduction
Combination antiretroviral therapy (cART) has significantly extended the life expectancy of people with HIV infection. However, HIV/AIDS is still one of the leading causes of morbidity and mortality worldwide. 1 Long-term drug-related toxicities as well as the emergence of multi-class drug resistant viruses highlight the continuing need for the development of new treatment options. 2 Raltegravir, a pyrimidinone analogue formerly known as MK-0518 and marketed as Isentress®, is the first inhibitor of the viral enzyme integrase approved by the US Food and Drug Administration (FDA) in October 2007 for the treatment of resistant HIV-1 in treatment-experienced patients in the first instance. It has been used effectively and safely in large international clinical trials in both treatment-naïve 3 and treatment-experienced patients with virological failure.4–6 Raltegravir along with other new antiretroviral agents such as the protease inhibitors (PIs) darunavir and tipranavir, the non-nucleoside reverse transcriptase inhibitor (NNRTI) etravirine and agents from 2 new classes, the fusion and entry inhibitors enfuvirtide and maraviroc will now allow clinicians to structure new treatment regimens with several fully active drugs with a high possibility of achieving long-lasting efficacy. This exciting opportunity is reflected in the recent update of the US Department of Health and Human Services (DHHS) treatment guidelines now recommending that viral suppression of plasma HIV-1 RNA to <50 copies/mL should be the aim of treatment even in treatment-experienced patients. 7
Mechanism of Action
Following entry of HIV-1 into the host cell cytoplasm, reverse transcriptase converts its single stranded RNA into double stranded DNA. Integrase then combines with viral DNA to form a stable complex containing viral and cellular proteins, known as the pre-integration complex which is actively transported to the cell nucleus. 8 Integration of HIV-1 DNA into the host cell genome is catalysed by the integrase enzyme in a two-step process. The first step involves the removal of two nucleotides from the 3’ ends of the HIV-1 DNA, followed by insertion of the HIV-1 DNA into the host cellular DNA widely referred to as strand transfer.9,10 The integrated viral DNA, termed proviral DNA serves as a template for virion production and subsequent infection of naive cells.11,12 Raltegravir inhibits the integrase enzyme thereby preventing the insertion of HIV-1 DNA into the host cell genome.13,14 Due to its novel mechanism of action it has been suggested that raltegravir will prevent the establishment of a viral reservoir for instance within resting CD4+ T cells when used early in the course of an HIV infection, resulting in an inhibition of ongoing HIV-1 replication and subsequent infection of new cells by competent virus. 15
Classically, the reduction in HIV replication after initiation of cART is characterized by a biphasic decay pattern of plasma HIV RNA until the viral load drops below the level of detection (50 copies/ml) of currently licensed clinical assays. 16 This pattern of plasma HIV RNA decay is consistent for reverse transcriptase and protease inhibitors. Recent data suggest that short term exposure to raltegravir as monotherapy as well as in combination with other antiretroviral drugs results in a substantially different pattern of plasma HIV RNA decay.3,17 The rate of HIV RNA decay is faster during the first phase and the second phase starts at an overall lower HIV RNA level. Several proposals based on early clinical studies have been put forward to explain the unique effect raltegravir has on the 2nd phase of viral decay.3,17–19 It is anticipated that current studies of raltegravir with frequent viral load measurements using a single copy viral load assay will shed more light on the viral decay characteristics. 15
Trial Results and Clinical Implications
A considerable number of phase I to III clinical trials has been conducted so far to assess the pharmacokinetic profile, efficacy, and safety of raltegravir along with examining its resistance profile and potential for drugdrug interactions. The following section summarises the key findings.
Pharmacokinetics
Results from the international, randomized, doubleblind, placebo-controlled, dose-ranging study known as Protocol 004 randomizing 35 antiretroviral-naïve HIV-infected patients to receive either placebo or raltegravir 100 mg, 200 mg, 400 mg, or 600 mg orally twice daily as monotherapy over a period of 10 days showed that mean trough minimum plasma concentrations were lowest in the 100 mg-dosing group and increased with dose (200 mg, 400 mg, 600 mg). 20
Mean maximum plasma concentrations and exposure area under the plasma-concentration curve (AUC) increased up to the raltegravir 400 mg dose but were similar in the 400 mg and 600 mg dosage study arms 20 supporting the future use of raltegravir at a dosage of 400 mg. Geometric mean plasma concentrations of raltegravir surpassed the mean in vitro 95% inhibitory concentration (IC95) for wild-type HIV-1 in the presence of 50% human serum (33 nM) in all 4 dosing groups.
Raltegravir is rapidly absorbed with a bioavailability of approximately 32%.21,22 Raltegravir is approximately 83% bound to human plasma proteins.
The primary clearance pathway for raltegravir in humans is hepatic glucuronidation mediated predominantly by the isoenzyme uridine diphosphate glucoronosyltransferase (UGT) 1A1,21,23 whereas NNRTIs, N(t)RTIs, and PIs are mainly metabolized by the cytochrome P450 enzymes. 21 At a concentration up to 100 μM, raltegravir had no significant effect on the cytochrome P450 system indicating that interactions with drugs metabolized by the CYP450 system are unlikely. 23
The apparent terminal t½ of raltegravir in humans is approximately 9 hours with a shorter α-phase half-life (~1 hour) accounting for much of the area under the curve (AUC). This pharmacokinetic profile supported the use of a twice-daily dosing regimen for raltegravir. 24 Of note, the currently recruiting phase 3 study known as QDMRK will compare once-daily vs twice-daily raltegravir therapy. With twice daily dosing, pharmacokinetic steady state is achieved within approximately the first 2 days of dosing.21,23,25 High fat meal slows the rate of absorption of raltegravir but also increases its exposure. Following a single 400 mg dose administration of raltegravir with high fat meal, the time to reaching maximum plasma concentration (Cmax) was delayed by 7.5 hours and the Cmax itself was decreased by 34%. However, the AUC was increased by approximately 19%. Therefore, raltegravir may be administered without regard to food. After an oral administration of radiolabeled raltegravir in healthy volunteers, approximately 51% was excreted in feces while 31% was excreted in urine. 21
No dose adjustment is required with respect to age, gender, body mass index, and HIV infection status.23,25 No dose adjustments are recommended in patients suffering from severe renal impairment or mild-to-moderate hepatic impairment.25,26
Raltegravir is currently classified as a category C drug allowing its use in pregnant women but only if the potential benefit outweighs the risk to the fetus. However, to date, no supporting data from clinical or pharmacokinetic studies exist. 27
Efficacy
Raltegravir in treatment-naïve patients
Results from 2 large clinical trials are available so far. Thirty participants from the initial Protocol 004 described above along with additional 171 were enrolled into the second part of the trial and randomized to one of five study arms receiving either efavirenz 600 mg once daily or one of the 4 raltegravir doses (100 mg, 200 mg, 400 mg, 600 mg orally twice daily) used in the first part of the trial twice daily over 48 weeks. 20 The 2 NRTI backbone regimen was the same for all 201 patients, tenofovir plus lamivudine. Patients were stratified by baseline plasma HIV-1 RNA < or >50,000 copies/ml. Raltegravir at all dosages showed a strong antiretroviral potency. 3 At week 4, ≥90% of all patients receiving raltegravir had reached <400 HIV-1 RNA copies/ml regardless of raltegravir dosage. Interestingly, among all 4 raltegravir dosage treatment arms at that timepoint 60% to 80% of patients had already reached <50 copies/ml compared with 25% of patients in the efavirenz arm which was statistically significantly earlier. 3 These findings generated substantial interest in the ability of raltegravir to influence the early phases of viral decay in a manner not witnessed following administration of any other antiretroviral drug.15,17 Differences in suppression rates vanished over time and 85% to 95% of patients across all 5 treatment arms achieved <50 copies/ml at weeks 24 and 48. Rates of virological failure were similar across all 5 treatment arms (n = 5 for the 4 RAL arms and n = 1 for the EFV arm) at 3%. 3 Additionally, no significant differences were observed in the mean increases in CD4+ T cell counts (range 144-221 cells/mm3) at weeks 24 and 48 between all treatment arms. 3
Week 48 data from the STARTMRK trial both confirmed raltegravir's antiretroviral potency and showed its virological non-inferiority to efavirenz. 28 In this blinded phase III study 563 treatment-naïve patients were randomised to either raltegravir 400 mg twice daily or efavirenz 600 mg once daily, both given with a fixed dose combination of tenofovir and emtricitabine once daily. Patients with genotypic resistance to efavirenz, tenofovir or emtricitabine at screening were excluded. 86% of raltegravir-treated patients achieved plasma HIV-1 RNA levels < 50 copies/ml at week 48 compared with 82% in the efavirenz arm which was statistically non-inferior (p < 0.001). 28 Additionally, fewer virologic failures occurred with raltegravir (10%) than with efavirenz (14%). Patients in the raltegravir arm experienced a significantly greater increase in CD4+ T cell counts over 48 weeks compared to patients in the efavirenz arm (189 cells/μl vs. 163 cells/μl, δ (95% CI) = 26 (4, 47)) as well as significantly faster decreases in plasma viral loads (p < 0.001). 29 Subgroup analyses also showed similar response rates among different viral subtypes. 90.3% HIV-1 clade B infected patients and 96.4% HIV-1 clade non-B infected patients receiving raltegravir achieved plasma HIV-1 RNA levels below 50 copies/ml at week 48. 29
Raltegravir in treatment-experienced patients
Results from 4 large clinical trials are available so far. An international, double-blind, phase IIb study called Protocol 005 examined the safety and efficacy of three different dosages of raltegravir (200, 400, and 600 mg orally twice daily) vs. placebo in 179 highly treatment-experienced HIV-1 infected patients. 4 The first 24 weeks were conducted double-blind after which patients were rolled over into an open-label phase in which all 100 continuing participants were given raltegravir 400 mg twice daily. Optimized background regimen (OBR) was chosen by the investigator on the basis of resistance testing results. Patients were stratified by use of enfuvirtide as part of their OBR as well as by their level of resistance to PIs. Neither darunavir nor tipranavir were allowed as part of the OBR. Patients were required to have experienced virological failure and acquired phenotypic and genotypic resistance to at least one drug in each of the three major ART classes (NRTI, NNRTI, and PI) at study entry. 33% to 38% of all patients across all treatment arms took enfuvirtide as an OBR drug and 40% to 57% had a phenotypic susceptibility score (PSS) of 0 to all then available antiretroviral drugs, i.e. in these cases not a single drug was left that was fully active against the patient's virus. A PSS of 0 to all PIs was confirmed in 84% to 98% of all 179 participants. Week 24 results revealed interesting changes to baseline in plasma HIV-1 RNA levels being the primary study endpoint. 56% to 67% of all raltegravir-treated patients reached <50 copies/ml compared to 13% in the placebo arm (p < 0.0001 for all RAL doses). 4 The most prominent increases in CD4 T cells counts were observed in the raltegravir 400 mg and 600 mg arms. Interestingly, around 90% of those patients who received enfuvirtide for the first time during this trial maintained plasma HIV RNA levels < 400 copies/ml at week 24.
Two large, international, phase 3 registrational clinical trials, BENCHMRK 1 and 2 (Blocking Integrase in Treatment Experienced Patients With a Novel Compound Against HIV-1: MERCK) have provided compelling evidence for raltegravir's antiretroviral potency in 699 treatment-experienced HIV-infected patients. 350 patients for BENCHMRK 1 were recruited in Europe, Peru, and the Asia/Pacific region, 349 patients were recruited for BENCHMRK 2 in North and South America. All were required to have evidence of virologic failure (HIV-1 RNA > 1,000 copies/mL) and documented resistance to at least one drug in each of the three ART classes. 6 Patients were randomised at a 2:1 ratio to receive raltegravir 400 mg or placebo twice daily with an optimized background regimen which was selected based upon previous treatment history and resistance testing results. The 400 mg twice daily dose was chosen taking into account the variability in individual raltegravir levels and the potential for drug-drug interactions with other antiretrovirals. 27 Analyses took place at week 16 (primary analysis) and week 48, both studies have been extended and will collect follow-up data until week 156. The permitted usage both of the fusion inhibitor enfuvirtide and of two then newly licensed ritonavir-boosted protease inhibitors, darunavir and tipranavir as part of OBR should be mentioned. 19% to 21% of all patients were using enfuvirtide for the first time, darunavir was given to 25% to 27% of patients in BENCHMRK 1 and to 45% to 50% in BENCHMRK 2. Around 90% of all patients had an AIDS defining illness in the past with an average duration of previous ART of 11 years. In BENCHMRK 1, 61% of raltegravir-treated patients vs. 33% in the placebo arm (p < 0.001) had reached plasma HIV-1 RNA levels <50 copies/ml at week 16, 60% vs. 34% at week 48. In BENCHMRK 262% of raltegravir-treated patients vs. 36% in the placebo arm (p < 0.001) had reached plasma HIV-1 RNA levels <50 copies/ml at week 16, 65% vs. 31% at week 48. Interestingly, if enfuvirtide or darunavir was part of the OBR for the first time patients were more likely to achieve virologic suppression by week 16. This rate is remarkable for patients infected with a triple-class resistant virus and is close to the rates seen in newly treated HIV patients. 30 Consecutive subgroup analyses from pooled data at week 48 also revealed that patients receiving raltegravir plus OBR achieved significantly better virological suppression across all CD4+ T cell count strata (≤50 cells/mm3, 50-200 cells/mm3, and ≥200 cells/mm3) than patients receiving placebo plus OBR. 5 The same applied to HIV-1 RNA strata (< and > 100,000 copies/ml). These analyses also showed similar response rates among different viral subtypes. 256 of 399 HIV-1 clade B infected patients (64%) and 24 of 36 HIV-1 clade non-B infected patients (67%) receiving raltegravir achieved plasma HIV-1 RNA levels below 50 copies/ml at week 48.
Data from the ANRS CO3 Aquitaine cohort confirm the efficacy of raltegravir plus OBR in highly pre-treated HIV patients in a routine clinical care setting. 31 73% of all patients (n = 51) had undetectable plasma HIV RNA (<50 copies/ml) at week 24.
Whilst the outcome from the BENCHMRK trials favored the use of raltegravir results from the SWITCHMRK studies were discouraging. The SWITCHMRK 1 and 2 trials were conducted in 702 treatment-experienced HIV-infected patients though without evidence of virological failure. Patients still had to have extensive triple class ART experience and had to be on ritonavir boosted lopinavir (LPV/r) plus 2 NRTI containing OBR for at least 3 months before being randomized to either remain on LPV/r (n = 352) or switch to raltegravir 400 mg twice daily (n = 352). 32 Both regimens included a placebo for the respective comparatordrug. 87% of all lopinavir-treated patients had undetectable viral loads (<50 copies/ml) at week 24 compared to 81% in the raltegravir arm in SWITCHMRK 1, 94% vs. 88% for SWITCHMRK 2. On the basis of these results a switch to raltegravir was classified as ‘not noninferior’ to staying with lopinavir. Further analyses revealed more NRTI and NNRTI resistance mutations in patients failing on raltegravir compared to patients failing on lopinavir/r potentially based on re-appearance of higher numbers of archived mutations in the raltegravir-treated patients along with a lower threshold for drug resistance of raltegravir compared to lopinavir/r.
On the other hand, results from 2 small non-randomized studies support the replacement of the salvage drug enfuvirtide with raltegravir in highly treatment-experienced patients on a stable regimen, around 95% of patients maintained undetectable plasma HIV-1 RNA levels at week 24.33,34
Interestingly, recent data support a dual therapy approach containing raltegravir and ritonavir-boosted darunavir in a salvage setting in highly treatment-experienced patients. 35
Results from the TRIO trial confirm the high rates of virologic success with co-administered raltegravir and ritonavir-boosted darunavir plus–-in this case–-etravirine in multiple class-experienced patients. 36 In this phase II non-comparative multi-centered study 90% of all patients (n = 103) had viral loads below 50 copies/ml at week 24.
Although not the focus of this review data on raltegravir's efficacy against HIV-2 infection have so far only been generated on an individual case level in highly treatment-experienced patients but deserve further investigations considering the low susceptibility of HIV-2 to currently licensed antiretroviral drugs. 37
In summary, raltegravir has shown significant antiretroviral efficacy in both treatment-experienced and -naïve patients38,39 even when compared to the current gold standard efavirenz that has been found to be superior to ritonavir-boosted lopinavir in the phase 3, randomized, open-label ACTG 5142 study. 41 Interestingly, patients with poor predictors to response to cART (high viral load, low CD4+ T cell count) also had greater efficacy with OBR plus RAL vs. OBR and placebo. 42 The results from the BENCHMRK trials have shown that viral suppression <50 copies/ml is now an attainable goal in highly treatment-experienced patients. Further studies will have to reveal if the observed altered viral decay rate translates into clinically significant findings.
Safety
Raltegravir in treatment naïve patients
Data from Protocol 004 indicate that total drug-related clinical adverse events at week 48 were more frequent with efavirenz (71%) than with raltegravir (48%). 3 85% of all adverse events across all 5 treatment arms were mild to moderate. No differences in numbers of serious adverse events could be observed between patients receiving raltegravir or efavirenz (5%-6%) and none of these were regarded to be drug-related or resulted in treatment discontinuation. In particular, no relationship between frequency of adverse events and the dose of raltegravir could be shown. Among the most common adverse events in the patients receiving raltegravir at any dose were nausea, dizziness, and headache. Neuropsychiatric events were more frequent in the study arm containing efavirenz at weeks 8 and 48 (21% vs. 8, and 29% vs. 13, respectively). One clinically interesting finding relates to the observation that raltegravir did not negatively influence serum lipids. Mean change from baseline to week 48 in total cholesterol for raltegravir-treated patients was -2.3 mg/dl (-0.06 mmol/L) vs. +20.7 mg/dl (+0.54 mmol/L) in the efavirenz arm which was found to be significant (p < 0.001). Additionally, significant differences between raltegravir- and efavirenz-treated patients could be shown for changes from baseline in levels of low density lipoprotein cholesterol and triglycerides (p = 0.016 and 0.07, respectively). 3 Levels of high density lipoprotein cholesterol rose in all 5 treatment arms from baseline to week 48, though to a lesser extent in patients receiving raltegravir vs. efavirenz (p = 0.01). Other grade 3/4 laboratory abnormalities which occurred generally at a low frequency in raltegravir-treated patients comprised decreased absolute neutrophil count, transaminitis, and increased pancreatic enzymes.
The favourable toxicity profile of raltegravir when compared with efavirenz could be confirmed in the STARTMRK trial. Overall, 90% of all raltegravir-treated patients and 96% of all efavirenz-treated patients (p = 0.002) experienced a clinical adverse event through 48 weeks. 28 Moderate to severe drug-related clinical adverse events (16% vs. 32%, p < 0.001) including CNS symptoms (10% vs. 18%, p = 0.015) were significantly less frequent in raltegravir-treated patients when compared to efavirenz-treated patients. Additionally, less grade 3/4 laboratory abnormalities were observed in the raltegravir arm, clear increases in total cholesterol and triglycerides in efavirenz patients in contrast to little changes in the raltegravir patients were present in STARTMRK over 48 weeks. Of interest, the total-to-high density lipoprotein cholesterol ratio stayed low in both treatment arms. 28
Despite being virologically ‘not noninferior’ switching to raltegravir was superior to staying with lopinavir in the SWITCHMRK trials with respect to lipid measures at week 12. 32 Decreases in total cholesterol, triglycerides and non-high-density lipoprotein cholesterol were noted in the raltegravir groups, whereas these parameters increased in the lopinavir groups (p < 0.001 for each comparison). Overall rates of clinical and drug-related adverse events were similar among the groups (70% and 13% in the raltegravir arms vs. 63% and 20% in the LPV/r arms, respectively).
Raltegravir in treatment experienced patients
The phase IIb study known as Protocol 005 revealed no raltegravir dose-related toxicities. 4 The most common drug-related adverse events occurring in at least 5% of all 45 patients treated with raltegravir 400 mg twice daily were vomiting (4.4%), nausea (4.4%), and diarrhoea (2.2%).
From both BENCHMRK trials overall clinical adverse events of any grade were reported in 89% of all patients receiving raltegravir vs. 87% in the placebo groups at week 16. 6 These were classified as treatment-related in 54% of all cases in all groups. 23% of all raltegravir-treated patients experienced laboratory adverse events, 22% in placebo arms, judged as related to study drugs in 14% and 13% of all cases, respectively. No further differences between study groups were seen at week 48. Similar to the treatment-naïve group, the most common side effects were diarrhea, nausea, and headache. Additionally, laboratory adverse events were related to increased serum lipid, aminotransferase, and creatinine levels. Importantly, raltegravir- vs. placebo-treated patients had a significantly higher percentage of increases (grade 4 = >20-fold) in creatinine phosphokinase (CPK) (2.2% vs. 0.7%, respectively). Against this background the FDA and the manufacturer recommend that raltegravir be used with caution in patients at risk for muscle problems; there have been rare reports of rhabdomyolysis in raltegravir-treated patients. 42
It has to be mentioned that week 16 results from both BENCHMRK trials suggested a higher risk of development of cancer among raltegravir-recipients. These were mostly HIV related, occurring in immunocompromised patients, of which some cases were recurrences of previously AIDS-related cancers. This early imbalance in rates of malignancies was no longer present at week 48 follow-up. 25 However, results from a recent review including a broad cancer definition and comprising findings from 5 randomized, double-blind, clinical trials of RAL in treatment naïve and experienced patients (Protocols P004, 005, BENCHMRK 1 and 2, and STARTMRK) and the open-label expanded access program (EAP-P023) showed no difference in risk of cancer in HIV-infected patients receiving RAL vs. other ARTs. 43
Results from 2 small non-randomized studies support the replacement of the salvage drug enfuvirtide with raltegravir in highly treatment-experienced patients on a stable regimen both from a virological and toxicity point of view, adverse events in 87 participants were infrequent and overall of mild to moderate grade, no new drug-related clinical or laboratory adverse events emerged.33,34 The authors speculate that a switch from an injectable to an oral antiretroviral drug and the subsequent disappearance of injection site reactions might enhance adherence to HIV medication.
Recently, four cases have been reported in which pre-existing depressive disorders exacerbated in treatment-experienced HIV-patients who newly started raltegravir and also were under treatment with antidepressants. 44 The authors hence recommend close monitoring in patients receiving ART who are under treatment with psychotropic medications and commence raltegravir.
In summary, raltegravir has exhibited a favourable safety profile in several large, international trials (see Table 1). Especially its neutral effect on serum lipids is of clinical interest in the light of emerging data on increasing numbers of cardiovascular diseases in longterm HIV-infected patients. Although attempts to switch patients from a fully suppressive regimen to a raltegravir-containing one because of e.g. an expectedly better metabolic profile should be undertaken cautiously in the light of the findings from the SWITCHMRK studies. However, follow-up data from large trials will need to confirm these encouraging findings on the long run.
Frequency of raltegravir plus OBR-related adverse events in patients from the Protocol 004, 005, startmrk and benchmrk trials.

•• common: >2%; • less common: ≤2% of patients receiving RAL plus OBR.
Resistance
To date, 25 antiretroviral drugs are approved for the treatment of HIV-1 infections divided into 6 categories, 45 more than 200 viral resistant mutations are associated with these antiretroviral drugs. 46 Whilst cART has been shown to control viral load, the virus is not eradicated and persists in latent reservoirs17,47 which allow continuing virus evolution and the subsequent development of drug resistant virus. 48 Another major contributing factor to virological resistance is non-adherence to the treatment regimen. Virological failure in treatment experienced patients due to the development of multi-drug resistance is a major problem in achieving complete viral control. Raltegravir with its novel mechanism of action might provide an attractive new treatment option in this setting.
Development of resistance to raltegravir has been reported in both antiretroviral-naïve and treatment-experienced patients in several large, international clinical trials: Protocol 004 and STARTMRK for ART-naïve and Protocol 005, BENCHMRK 1 and 2, and SWITCHMRK for treatment-experienced patients),3–5,29,32 which has provided valuable insight into the mechanism of resistance to raltegravir. Three mutational pathways occurring under selective antiretroviral pressure have been identified so far leading to partial or full resistance to raltegravir (see Table 2).
Raltegravir-related resistance pathways.
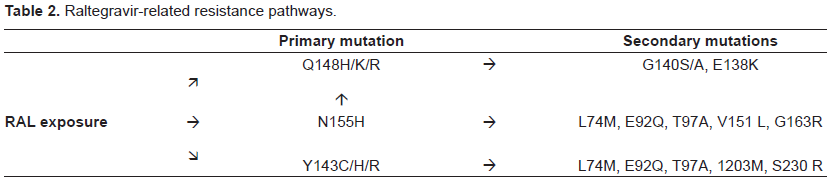
Raltegravir resistance is primarily associated with mutations at positions 148 (Q148H/K/R) or 155 (N155H) of HIV integrase which do not occur together. However, each of these primary mutations usually occurs together with one or more secondary mutations. 49 The primary mutation at position 155 is associated with several secondary mutations including L74M, E92Q, T97A, V151L and G163R. Secondary mutations linked to codon 148 include G140S/A and E138K. Mutations with different secondary mutations associated with the Q148 resistance pathway confer higher resistance than the N155 pathway. 14 Results from the BENCHMRK studies 1 and 2 revealed that in treatment-experienced patients with virological failure a high level of resistance was associated with raltegravir 64/94 (68%), of these 26 had a mutation at codon 148 and 38 at codon 155. 5 However, a number of other studies indicate that the Q148 pathway is more frequently associated with resistance to raltegravir in patients experiencing virological failure than the N155 pathway.14,41,50 It has been reported that in some raltegravir-treated patients resistance profiles switched from the N155 pathway to the Q148 pathway after a few weeks. 51 The Q148H mutation mostly occurs together with the G140S mutation. The Q148H mutation may be present as a single mutation at the start of raltegravir treatment, but the G140S mutation usually appears after a few weeks of treatment. The G140S/Q148H double mutation confers strong resistance to the drug leading to viral replication levels similar to those of the wild type (WT) virus. Importantly, the G140S displays weak resistance (IC50 = 30 nM) while the Q148H is strongly resistant to RAL (IC50 > 700 nM). All mutants of the integrase gene, including G140S, displayed replication kinetics similar to those to the WT virus, with the exception of the Q148H mutant which is characterized by a slower kinetic. Consistently, all mutants are weakly impaired in the synthesis of viral DNA as well as in their propensity to integrate their genome and consequently to produce viral particles 52 with the exception of the Q148H mutant. Its integration efficiency is 7-fold less in comparison to the WT. These data demonstrate that, in the case of the G140S/Q148H double mutant, the resistance to raltegravir is due to the Q148H mutation and that the G140S mutation rescues the integration deficiency and thus the kinetic of replication. 53
A third and less common pathway associated with raltegravir resistance involves a mutation at position 143 (Y143C/H/R) and is associated with mutations at L74M, E92Q, T97A, 1203M and S230R.5,14,54
A switch from the N155 pathway to the less common Y143 pathway has been observed in a small number of patients receiving raltegravir plus OBR in a French cohort.55,56
In the absence of the three primary resistance pathways the E92Q mutation was observed with T66 A followed by L741. 57 A few patients in the BenchMRK studies with virological failure exhibited a mutation at E92Q only. Others mutations, such as the E92 and E157Q, have also been described. 58
The integrase gene is highly variable with natural polymorphisms commonly occuring but primary mutations associated with resistance to integrase inhibitors are rare (<0.5%).58,59 A Brazilian study reported the occurrence of natural genetic polymorphisms in the integrase gene among HIV-1 subtypes B, C, and F but did not observe any of the primary mutations associated with raltegravir. 60 One of the primary mutations associated with resistance to raltegravir N155H was detected in only 1.5% of samples of antiretroviral-naïve patients. 61 Another study by Eshleman and colleagues conducted in antiretroviral-naïve patients found that 8.3% exhibited mutations associated with reduced susceptibility to integrase inhibitors and these varied according to HIV subtype. 62 A polymorphism at position T206S at baseline has been described to be potentially related with resistance to raltegravir.31,55 Ceccherini-Silberstein and colleagues suggest that natural polymorphisms evident prior to raltegravir therapy may influence the emergence of primary resistance mutations to raltegravir. 58
There is evidence of cross-resistance between raltegravir and elvitegravir, an integrase inhibitor not yet licensed. Mutations under elvitegravir occur at E92Q, Q148R/K/H and N155H14,63,64 suggesting a common mechanism in developing resistance to both these integrase inhibitors. 54 This curtails the therapeutic approach of sequencing of integrase inhibitors. In vitro studies show that viruses harbouring a number of integrase mutations were not only resistant to raltegravir and elvitegravir but also to a number of additional integrase inhibitors still in development. 54 These findings suggest that future development of non cross-resistant second generation integrase inhibitors will be difficult. 46
Importantly, most studies examining the genetic polymorphism of integrase and the natural susceptibility of HIV to integrase inhibitors have focused on individuals infected with HIV-1. A recent study in 52 treatment-naïve HIV-2 infected patients from the French HIV-2 cohort ANRS CO 05 examined the polymorphism of the integrase gene. 65 Despite 40% heterogeneity between HIV-1 and HIV-2 integrase genes, phenotypic susceptibility to the integrase inhibitors raltegravir and elvitegravir was found to be similar to that observed for HIV-1. The conclusion that integrase inhibitors may represent a therapeutic option for HIV-2 infected patients has recently been challenged by a case report showing development of N155H in an HIV-2 infected patient. 66
In summary, functional monotherapy with raltegravir should be avoided due to its low genetic barrier to the development of resistance. This highlights the need for a wisely chosen, strong OBR as reflected in the lower rate of virological failures in the BENCHMRK trials compared to Protocol 005 in which the newly licensed ritonavir-boosted protease inhibitors darunavir and tipranavir were not allowed as part of the OBR.
Drug interactions
Raltegravir is metabolized by hepatic glucuronidation via the enzyme UGT1 A1 and not like most other antiretroviral agents by the cytochrome P450 pathway offering the promising potential for minimal drugdrug interactions between antiretroviral drugs as well as other concomitant medications.25,67 Additionally, recent longitudinal data confirm that current RT or PI containing regimens do neither reduce the fitness of viral integrase nor affect raltegravir efficacy. 68 Not only does raltegravir not inhibit or induce cytochrome P450 enzymes, 69 it has also been shown to have no inhibitory effect on P-glycoprotein-mediated drug transportation. 25 This is a clinically important finding as it suggests that raltegravir might not significantly influence pharmacokinetics of frequently co-administered drugs such as statins, azole antifungals, proton pump inhibitors, opioids, methadone, oral contraceptives, and erectile dysfunction therapies.26,39,70
However, drugs inducing or inhibiting UGT1 A1 may alter raltegravir plasma levels. To date, limited data are available for a number of antiretroviral drugs as well as concomitant medications, further studies are needed to unravel raltegravir's full interactive profile (see Table 3).
Currently known effects of antiretroviral and concomitant medications on raltegravir plasma levels.
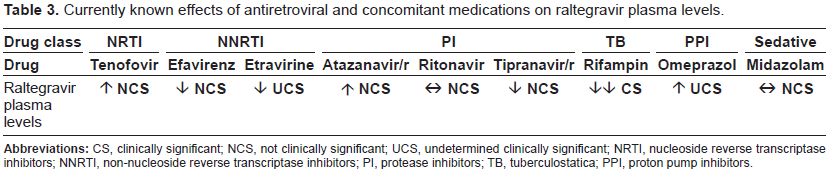
Atazanavir is a potent inhibitor of UGT1A1 and was therefore selected to assess its effect on increases in raltegravir plasma levels. 77 Both atazanavir alone and in combination with ritonavir were investigated in phase 1 studies. Additional data on this interaction was also obtained from the phase 2 population PK data. In those studies, raltegravir plasma levels were increased when co-administered with atazanavir, consistent with inhibition of UGT1A1. The increases, however, were on the whole modest (30%-70% increases in AUC) and not considered clinically meaningful. 71 Concomitant use of raltegravir and atazanavir was well tolerated in the phase 2 and 3 studies. Based on these data, atazanavir may be coadministered with raltegravir without adjustment in the dose of raltegravir.
Another antiretroviral drug, the protease inhibitor Tipranavir reduces raltegravir plasma levels by inducing UGT1A1. 25 But findings from a study conducted in 100 HIV-infected patients treated with raltegravir plus either ritonavir-boosted tipranavir or a comparator PI confirmed similar efficacies for both regimens. 72
Co-administration of raltegravir with tenofovir leads to increases in plasma levels of both drugs, by 49%-64% for raltegravir and by 10%-13% for tenofovir. 73 Again, these findings were reported to not be clinically significant.
Additional studies have also confirmed lower though clinically not significant raltegravir plasma levels when co-administered with efavirenz, 74 and etravirine. 75 Although for the latter, four recently reported cases have shown significant decreases in raltegravir trough concentrations when co-administered with etravirine in HIV-infected patients. 76 Against this background the authors highlight the need for therapeutic drug monitoring (TDM) in this setting at least until data from large cohorts are available.
No changes were observed when ritonavir was co-administered. 74 This might put a protease inhibitor-boosting strategy in the optimization of therapeutic raltegravir plasma levels into perspective, i.e. when pursuing once daily dosing.
When discussing drug-drug interactions one needs to pay special attention to antibacterial drugs used in the treatment of tuberculosis. One frequently used agent in this setting is rifampin. It should be used with caution in raltegravir-treated patients as it has been found to reduce plasma levels of raltegravir by 38%-61%. The manufacturer recommends double dosages of raltegravir with 800 mg twice daily to achieve therapeutic plasma levels. 25
Additionally, a small study comparing pharmacokinetic parameters of raltegravir plus the sedative midazolam vs. midazolam alone in 10 healthy volunteers revealed no clinically significant interactions. 77
Patients receiving ART who are also under treatment with psychotropic medications and commence raltegravir should be closely monitored on the basis of four recent case reports indicating exacerbations of depressive disorders in treatment-experienced HIV-patients who newly started raltegravir. 44
Lastly, due to a disproportionate increase of serum CPK levels in treatment-experienced patients in the study arms containing raltegravir vs. placebo of the BENCHMRK trials the FDA and the manufacturer recommend that raltegravir be used with caution in patients at risk for muscle problems; there have been rare reports of rhabdomyolysis in raltegravir-treated patients. Concomitant medications known to cause this condition should be administered with extreme care. 42
Conclusions
Raltegravir has demonstrated impressive antiretroviral potency in both treatment-experienced and -naïve patients in several large, international trials. It appears to have a favorable safety profile and since it is metabolized via the UGT1 A1 pathway less frequent drug-drug interactions were observed. However, its low genetic barrier to resistance development highlights the need for a wisely chosen, strong background regimen, especially as in the event of virological failure sequencing of integrase inhibitors does not seem to be an option with regards to emerging data on cross resistance. Overall, raltegravir will be a valuable addition to the antiretroviral treatment arsenal and has, together with other recently approved antiretroviral drugs, the potential to alter the prevailing treatment paradigm of combining two NRTIs with a PI or an NNRTI as the corner stone of combination antiretroviral therapy.
Disclosures
The authors report no conflicts of interest.