
Abstract
Pulmonary arterial hypertension (PAH) is defined as a group of diseases characterized by a progressive increase in pulmonary vascular resistance (PVR) leading to right ventricular failure and premature death. Untreated, it is a potentially devastating disease. However, the past decade has seen remarkable improvements in our understanding of the pathology associated with the condition and the development of multiple PAH-specific therapies with the ability to alter the natural history of the disease. These new advances provide a significant opportunity for practitioners to detect and treat patients with PAH in a timely and effective manner, thereby improving overall mortality, morbidity, and quality of life associated with this disease. The aim of this review is two-fold: firstly to review the evidence for efficacy and safety of non-parenteral PAH therapies and to discuss treatment selection based on clinically meaningful differences among the approved therapies, such as the potential for serious drug-drug interactions, convenience of dosing schedules, and rates of limiting side effects. Secondly, the central role of the PAH clinical nurse in the multidisciplinary care of patients with PAH will be discussed, together with issues relating to adherence and interventions to enhance patient compliance.
Keywords
Introduction
Pulmonary arterial hypertension (PAH) is a group of diseases that affect the small pulmonary arteries, and which form a subset of those with pulmonary hypertension (PHT). It is a devastating and life-threatening disease characterized by elevated levels of endothelin-1 (ET-1), cell proliferation and hypertrophy, in situ thrombosis and deposition of fibrotic tissue.1,2 This process causes an increased resistance to the flow of blood through the lungs, resulting in raised pulmonary arterial pressure (PAP) against which the right ventricle must work. Patients with PAH commonly develop progressive right ventricular (RV) hypertrophy and dysfunction, with most ultimately succumbing to right heart failure. 3 Associated symptoms, which include dyspnea, chest pain and fatigue, become progressively debilitating, severely limiting activities of daily living, particularly in the later stages of the disease. 4
Until the early 1990's, mortality rates in patients with PAH were high: the median life expectancy of idiopathic PAH (iPAH) without specific therapy was 2.8 years from diagnosis, with 1-year, 3-year, and 5-year survival of 68%, 48% and 34%, respectively. 5 However, the field of PAH has been advancing rapidly in recent years, and that pace continues to accelerate in respect to developing effective therapeutic options. A number of pharmacological agents have been approved based on positive results from randomized clinical trials (RCTs), namely prostanoids (epoprostenol, iloprost and beraprost), a phosphodiesterase-5-inhibitor (PDE-5, sildenafil), and ET-receptor antagonists (bosentan, sitaxsentan and ambrisentan). All of these agents have the ability to favourably alter the natural history of PAH. A recent meta-analysis of RCTs in PAH, suggested that active treatments were associated with a reduction in mortality of 43% [relative risk (RR): 0.57; 95% confidence interval (CI): 0.35 to 0.92; P = 0.023) and a 61% reduction in the incidence of hospitalizations (RR: 0.39; 95% CI: 0.25 to 0.61; P < 0.001). 6
Despite clear gains in the therapeutic armoury to tackle PAH, the selection of the most appropriate therapy is complex and problematic, and requires familiarity with the disease process, evidence from treatment trials and consideration of a range of practical issues; including potentially complicated drug delivery systems, dosing regimens, side effects, and adverse effects. This article aims to review the non-parenteral treatment options for PAH, objectively detailing the evidence available to support each form of therapy. In addition we describe problems encountered in daily practice that may complicate patient management such as drug-drug interaction, tolerability, and issues relating to patient adherence.
Methods
We conducted literature searches limited to randomized placebo-controlled trials published in English in MEDLINE and EMBASE (1966–August 1, 2010) using the search terms bosentan, ambrisentan, sitaxsentan, sildenafil, iloprost and beraprost (non-parental forms of treatment for PAH tested in RCTs). Full articles were used in lieu of abstracts when available. We considered studies conducted among patients with known or suspected iPAH or PAH occurring in association with underlying connective tissue disease (CTD), or congenital heart disease (CHD). We excluded studies of PHT associated with chronic thromboembolic disease, chronic obstructive pulmonary disease (COPD) or other parenchymal lung disease, high-altitude PHT, or cardiac disease (eg, left-heart failure, valvular heart disease) except congenital heart disease. Although intravenous epoprostenol is widely considered to be the most potent treatment for PAH, its use will not be considered here. Despite its benefits, the complexity of drug administration limits its use, 7 and it is often reserved for those patients with World Health Organization (WHO) functional class IV symptoms or patients refractory to first- and second-line therapy.8,9
Results
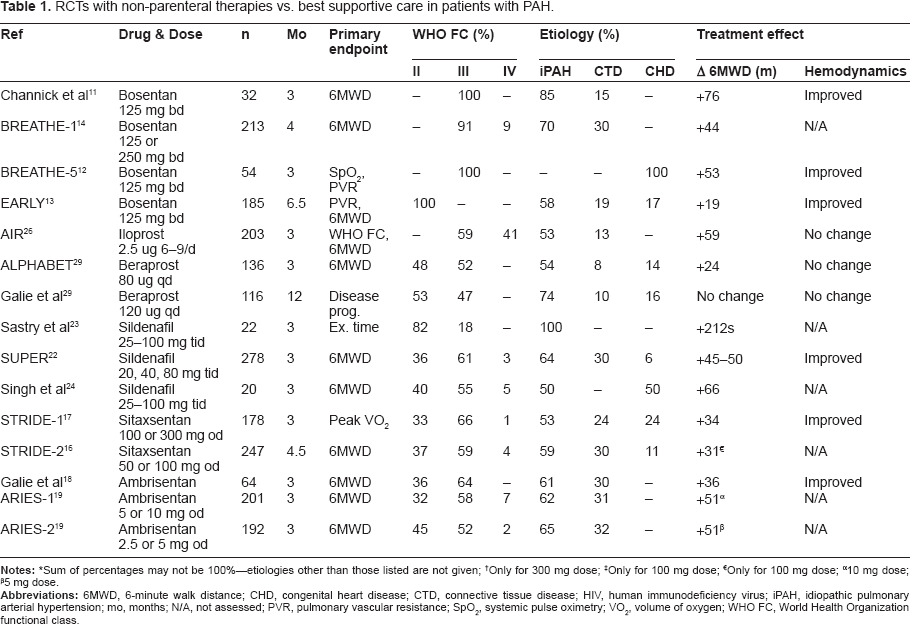
A summary of results is also presented in Table 1.
RCTs with non-parenteral therapies vs. best supportive care in patients with PAH.
Only for 100 mg dose;
10 mg dose;
5 mg dose.
Treatment options
ET-receptor antagonists
In patients with PAH, ET-1 levels are increased and correlate with disease severity.9,10 ET-1 causes vasoconstriction of pulmonary vessels and stimulates smooth muscle and fibroblast proliferation by acting at ET-receptor subtypes A and B. Three ET-receptor antagonists are currently available and discussed in this review: bosentan, sitaxsentan and ambrisentan.
Bosentan is an oral, dual ETA and ETB-receptor antagonist. Activation of ETA and ETB-receptors on smooth muscle cells mediate the vasoconstrictive and mitogenic effects of ET-1. The efficacy and safety of bosentan has been extensively studied in four RCTs.11–14 The pivotal and largest trial, Bosentan Randomized Trial of Endothelin Antagonist Therapy (BREATHE-1), confirmed the benefits of bosentan given at a dose of 125 mg twice-daily in improving the distance walked in six minutes (6MWD), cardiopulmonary hemodynamics, Borg dyspnea index, and WHO functional class, whilst increasing the time to clinical worsening. 14
Positive data regarding the use of bosentan also extend to PAH associated with Eisenmenger's syndrome (BREATHE-5 trial), 12 and patients with WHO functional class II symptoms (EARLY trial). 13 Based on the consistency and robust nature of these results, current PAH guidelines assign bosentan a level A recommendation in patients with WHO functional class II and III symptoms. 8
Sitaxsentan and ambrisentan are both orally active, ETA selective antagonists. Sitaxsentan has been assessed in WHO functional class II–IV PAH patients in two RCTs (Sitaxsentan to Relieve Impaired Exercise, STRIDE 1 and STRIDE 2).15–17 Both trials demonstrated improvement in exercise capacity (assessed by the 6MWD). Unlike the studies with bosentan, however, there was no difference seen in the time to clinical worsening, and in one of the two studies, 17 the primary end point (peak oxygen consumption as assessed by cardiopulmonary exercise testing) was not statistically significant. Ambrisentan has been evaluated in three RCTs.18,19 The results of the Ambrisentan in Pulmonary Arterial Hypertension, Randomized, Double-Blind, Placebo-Controlled, Multicenter Efficacy Studies (ARIES-1 and ARIES-2) comprise the most clinically meaningful data set for this drug. Together, they demonstrated the efficacy of ambrisentan in improving exercise tolerance in WHO functional class II and III patients without any major safety concerns. Variable benefits were seen in time to clinical worsening and improvement in WHO functional class, although these studies were not powered to properly determine drug effect on these parameters.
PDE-5 inhibitors
Sildenafil is an oral PDE-5 inhibitor. PDE-5 inhibition exerts its pharmacological effect by decreasing the breakdown of endogenous nitric oxide available, thereby inducing relaxation and anti-proliferative effects on vascular smooth muscle cells. 20 Early clinical studies with sildenafil in patients with PAH have demonstrated its ability to reduce mean pulmonary artery pressure (mPAP) and pulmonary vascular resistance (PVR), and to produce an increase in cardiac index (CI). 21
Three RCTs with sildenafil as monotherapy versus best supportive care have been performed in patients with PAH.22–24 In those trials, the dose of sildenafil ranged from 60 to 240 mg/day. In two trials,23,24 the methodology was relatively poor, patient numbers were low and the investigators employed unique doses of sildenafil. The results from those trials should be treated with some caution. In the largest RCT performed by Galie et al, 22 278 patients were randomized to receive placebo or sildenafil (20, 40 or 80 mg) orally three-times daily for a period of 12 weeks. The primary efficacy endpoint was the change in 6MWD from baseline to week 12. Compared to placebo, a significant increase in 6MWD was observed in all three sildenafil groups: 45 m (P < 0.001), 46 m (P < 0.001) and 50 m (P < 0.001) for sildenafil 20 mg, 40 mg and 80 mg, respectively. Moreover, there was no statistically significant difference in treatment effect between sildenafil doses. However, the same cannot be said for the observed improvement in hemodynamics and changes in WHO functional class. The effects on these parameters did appear to be dose dependant and patients randomized to a higher dose of sildenafil demonstrated greater improvements in mPAP, CI, PVR, and WHO functional class status.
Prostacyclin analogs
Iloprost is a prostacyclin derivative and was first approved by the US Food and Drug Administration (FDA) in 2004 for WHO functional class III and IV PAH. Prostacyclin is a metabolite of arachidonic acid produced primarily by the vascular endothelium. It is a potent vasodilator, affecting both the pulmonary and systemic circulation and there is evidence to suggest that a relatively deficiency of prostacyclin may contribute to the pathogenesis of PAH. 25 Short-term data for inhaled iloprost as monotherapy was available from one RCT that enrolled patients with both PAH and chronic thromboembolic pulmonary hypertension (CTEPH). 26 Overall, that study showed an increase in exercise capacity, an improvement in PVR, and clinical events for patients receiving iloprost. However, other hemodynamic measures were not affected.
A second prostacyclin analog, beraprost sodium, is also available for use in patients with WHO functional class II or III PAH. 27 While not currently approved for use in the United States or Europe, it is approved in Japan and several other countries. 28 In a 12-week RCT involving 130 patients with PAH [all in New York Heart Association (NYHA) functional class II or III] caused by various conditions (including iPAH, CTD-PAH, CHD-PAH and HIV-PAH), 29 patients who received beraprost (at a median dose of 80 μg, four-times daily) had a mean increase of 24 m on the 6MWT, and patients with iPAH had a mean increase of 46 m (P = 0.04 for both comparisons). Patients with other forms of PAH had no significant changes in their exercise capacity. In the overall study population, the administration of beraprost did not significantly change cardiovascular hemodynamics.
A longer, 12-month RCT confirmed that patients in NYHA functional class II or III who were treated with beraprost had improved scores on the 6MWT at three months and six months, as compared with the standard therapy group. However, this effect was not sustained at nine and 12 month assessments. 27
Dosage and administration of pharmacological agents
A summary of recommended dosing schedules for the approved non-parental PAH therapies is presented in Table 2. Sitaxsentan and ambrisentan are administered once-daily, while bosentan and sildenafil are administered twice, and three-times daily, respectively. Iloprost is administered via an adaptive aerosol device with a half-life of 20 to 25 minutes. Therefore, chronic use requires six-to-nine inhalations per day to obtain a sustained clinical benefit. With a jet nebulizer, the duration of each inhalation takes approximately 15 minutes; and with alternative devices such as ultrasound nebulizer, the inhalation time can be reduced to five minutes. 30 From a practical perspective, the administration schedule (2.5 μg or 5 μg six-to-nine times/day), and the fact that the hemodynamic effects disappear within 30 to 90 minutes, limit the attractiveness of iloprost for many patients.
Summary of product characteristics.
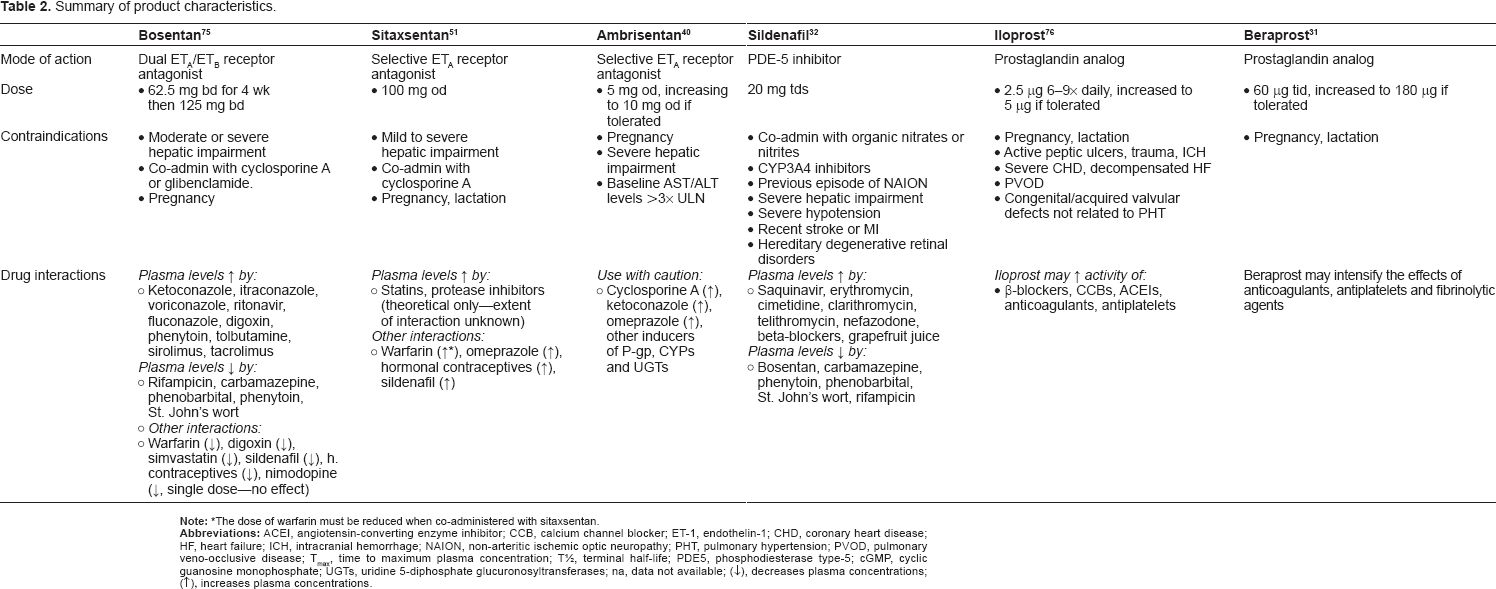
Beraprost is administered orally and should be taken after meals. Treatment is initiated at 60 μg divided into three doses. 31 The dosage can be increased gradually under careful monitoring up to a maximum dosage of 180 μg daily divided in three-to-four doses. However, in patients enrolled in the 12-week ALPHABET trial, the dosage was increased from 20 μg four times a day up to a mean and maximum dosage of 80 and 120 μg four times a day, respectively. 29
The approved dose of sildenafil in patients with PAH is 20 mg three times-daily, administered orally. 32 However, there are some caveats with this recommendation that require consideration. Given the dose-dependent observations in cardiopulmonary hemodynamics with sildenafil in the SUPER trial, 22 and the prognostic importance of hemodynamics on patient outcomes,5,33–35 a higher dose of sildenafil (eg, 80 mg three times-daily) may be warranted in the majority of patients.
To date, no studies have assessed the effects of dosage regimes on patient adherence in PAH. However, for many chronic diseases, research has shown that adherence decreases as the complexity of the medication regimen increases. 36 In a general review of chronic disease states, Claxton et al, identified 76 studies conducted between 1986 and 2000 which used electronic monitoring systems to assess adherence. Compliance with once-daily, twice-daily, three times-daily and four times-daily dosing requirements was assessed. The authors defined two major categories of compliance: dose taking (taking the prescribed number of pills each day) and dose-timing (taking pills within the prescribed time frame). The mean dose taking compliance estimated from these studies was 71% (range 34%–97%). Adherence rates declined as the number of daily doses increased: one dose 79% ± 14%, two doses 69% ± 15%, three doses 65% ± 16%, four doses 51% ± 20% (P < 0.001 among dose schedules). Compliance was significantly higher for once-daily versus three or four times-daily regimens; however, there were no significant differences in compliance between once-daily and twice-daily regimens or between twice-daily and three times-daily regimens. The authors concluded that simpler, less frequent dosing regimens resulted in better compliance across a variety of therapeutic classes of medicines. Whether these results can be generalized to the PAH setting remains to be confirmed.
Safety and efficacy
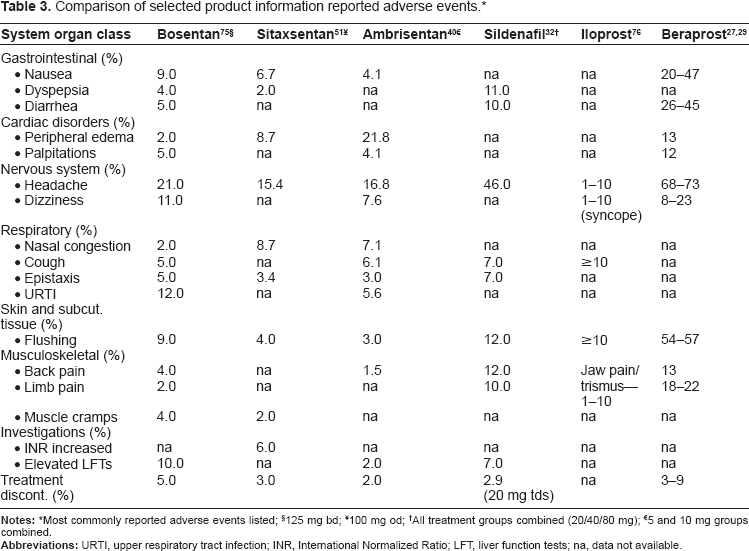
Although the non-parenteral therapies for PAH are generally well tolerated, they are associated with side effects related to their vasodilatory properties including peripheral edema, headache, and flushing. Table 3 provides an overview on the incidence of those side effects that have the greatest relevance in everyday clinical care.
Comparison of selected product information reported adverse events. *
§125 mg bd;
¥100 mg od;
†All treatment groups combined (20/40/80 mg);
€5 and 10 mg groups combined.
Hepatotoxicity
The most clinically relevant side effect reported with ET-receptor antagonists is dose-dependent liver function abnormalities. These present as elevated transaminases and/or bilirubin levels, and because these changes are a marker for potentially serious liver injury, serum aminotransferase levels (and bilirubin if aminotransferase levels are elevated) must be measured prior to initiation of treatment and monthly thereafter. It has been reported that bosentan inhibits the bile salt export pump, which may lead to cholestatic liver injury as a result of the intracellular accumulation of bile salts, while increasing bile salt-independent bile flow.37,38
While the incidence of hepatotoxicity in the RCTs was highest with bosentan (Table 3), the inclusion criteria and control group characteristics of the studies should be taken into account. Patients with liver enzyme elevations >1.5x upper limit of normal (ULN) at baseline were excluded from the ambrisentan studies (ARIES-1, ARIES-2), 19 and the STRIDE-2 study with sitaxsentan. 16 Meanwhile, patients with elevations up to 3x ULN were included in STRIDE-1 (sitaxsentan) 17 and BREATHE-1 (bosentan). 14 Similarly, the incidence of hepatic aminotransferase elevations >3x ULN in the control groups varied between 0 and 6%.
Consequently, drug surveillance programmes (notably, the TRAcleer eXcellence Post-Marketing Surveillance Programme, TRAX PMS) for bosentan, have been established to obtain safety data from real-life usage. 39 Seventeen countries participated in the TRAX PMS, enrolling 4,994 patients with PAH. Moreover, 4,623 (93%) were naïve to bosentan at study entry. Over 30 months, elevated aminotransferases were reported in 352 (7.6%) patients and only 150 (3.2%) patients discontinued bosentan as a result. Liver function monitoring is recommended in all patients receiving ET-receptor antagonists. However, the results of TRAX PMS suggest that elevated liver aminotransferases can be effectively managed in the majority of patients. The ongoing assessment of monthly blood tests is inherent in PAH management, however, no formal evaluation of these blood tests have been carried out in relation to patient adherence to ERA's in PAH.
Peripheral edema
In the RCTs performed to date, peripheral edema has been observed in up to 17% of patients receiving ambrisentan. 40 In addition, a warning label has been issued by the FDA for ambrisentan based on post-marketing reports of fluid retention occurring within weeks of starting therapy. 41 Although there has been speculation as to whether peripheral edema occurs more frequently as a drug-specific, ETB-mediated side effect, a closer examination of the incidences of leg edema in the pivotal studies with ET-receptor antagonists suggest that the incidence is related to patient characteristics (as can be derived from the large variance in the placebo groups), and do not suggest a significant drug-related effect.
Drug-drug interactions
Bosentan, ambrisentan, and sitaxsentan display clinically important differences with respect to drug interactions, and their potential for use in combination therapy (Table 2). Of interest is the interaction of bosentan with sildenafil, a frequently used combination, where sildenafil plasma levels are reduced by about 50% while bosentan concentrations rise by approximately 50%.42,43 Theoretically, sub-therapeutic sildenafil levels as well as increased bosentan-related liver toxicity may result. However, in clinical practice, this combination is well tolerated and appears to be effective.44–49
Another important concomitant medication in patients with PAH is warfarin. Bosentan and sitaxsentan have different effects on the doses of warfarin: bosentan partially induces the cytochrome P450 system, thereby increasing warfarin metabolism and the required dose. 50 In contrast, sitaxsentan inhibits the liver isoenzyme CYP2C9. Thus, combining sitaxsentan and warfarin in healthy volunteers can lead to a 2.4-fold increase in exposure to warfarin, therefore, requiring a substantial reduction in dose (~80%) at initiation of therapy to avoid bleeding complications. 51 No such interaction occurs with ambrisentan; however, according to the product labelling, the drug interaction potential of ambrisentan ‘has not been well characterized’. 40
Discussion: The Clinical Nurse Perspective
PAH is clearly a chronic, life-shortening disease and many patients suffer from limitations in their physical mobility, energy, emotional reactions and social isolation. As described above, the treatment and management of PAH is both complex and ongoing. Understandably, therefore, many patients also experience progressive worsening of their condition because they are non-adherent with the requirements of their health care regimen. Regardless of the type of therapy employed, therefore, clinical nurses with an interest and (ideally) specialist training and skills in PAH are in an ideal position to assess predictors of non-adherence as well as to implement interventions to enhance patient compliance.
In many Centers of Excellence in PAH, the clinical nurse plays a central role in the multidisciplinary management of patients with PAH. Indeed, the effective diagnosis and treatment of PAH, where several clinical pathways and scenarios are possible, relies on the effective coordination of many clinical specialties with the knowledge and skill to meet the holistic needs of the patient and their carers.52,53 Clinical nurses use expert knowledge, skills, and reasoning to direct and manage the care of patients within their specialty. Their scope of practice includes direct patient care, consultation, research utilization, and education. 54 More specifically, the role of the clinical nurse in the management of patients with PAH includes:
Identification and initial work-up of potential PAH patients.
Organisation of initial screening examinations including echocardiography, ECG analysis and chest x-rays.
Coordination of multidisciplinary team meetings.
Patient education and consultation during hospitalization, outpatient visits, and telephone follow-up.
Through this close relationship with the patient, the PAH clinical nurse is able to identify key factors leading to non-adherence and tailor interventions to prevent complications.
Predictors of non-adherence with treatment medication include, but are not limited to, the patient's level of education,55,56 comorbidity, 57 number of pills taken per day, 58 frequency of medication, 59 poor relationships and a lack of social support.60–62 In addition, treatment-related factors such as the patient's barriers to medication adherence (eg, forgetting the time of medication; a belief that skipping one dose is okay), also need to be considered. 63 The Clinical Nurse is often the first person contacted in the multidisciplinary team and receives most of the initial enquiries when there are problems with medications. A critical part of the PAH clinical nurse's role is to identify these predictors early and, when appropriate, initiate interventions to improve adherence. This may include an assessment of the patient's beliefs about the importance of adhering to the therapeutic regimen, repeating and reinforcing patient education, facilitating the adjustment of medications to minimize side effects, dietary counselling, and activity monitoring. In this regard, the building of rapport and development of a quality relationship with the patient is essential but often overlooked. A trusting relationship can create opportunities for full disclosure of the patient's feelings and expectations that can continue throughout the patient's course. Having a blended background of clinical training and holistic beliefs, the PAH clinical nurse is able to foster this type of relationship.
The use of counseling and education are important strategies in the care of PAH patients, and there is a growing body of literature to demonstrate that patients receiving nurse-led case management are more likely to receive interventions that are inline with evidence-based treatment guidelines.64–66 Furthermore, it seems clear that when nurses work in close partnership with patients–as in nurse-led multidisciplinary teams–patients are more responsive to treatment regimens, possibly due to being able to discuss problems and understanding the importance of medication adherence. 67
There is no comprehensive information describing how, or to what extent, a nurse-led multidisciplinary approach to PAH management affects patient outcomes. However, the effectiveness of such a coordinated approach has been well documented in other chronic conditions, namely heart failure,54,68–72 stroke, 73 and oncology. 74 A quantitative assessment of long-term patient outcomes in the PAH setting represents a potential avenue for future research.
Conclusions
The optimal therapy for patients with PAH is a highly individualized decision, taking into account many factors including, drug availability, severity of illness, route of administration, side effects, treatment goals, and clinician preference. Considering the entire group of PAH patients who have been prospectively studied in RCTs with non-parenteral therapies, very few clinically meaningful differences between agents can be demonstrated to date. The exceptions to this are iloprost and beraprost, where efficacy and compliance issues are more suspect, and to a smaller extent sildenafil which, from a clinical perspective, may require higher dosing to achieve the intended therapeutic effect. For the other agents discussed in this review (ie, bosentan, sitaxsentan and ambrisentan), much longer follow-up periods and probably other endpoints might be necessary to detect clinically important differences. Until the results from such long-term trials become available, other features are likely to be of greater relevance when considering treatment, such as clinical experience, convenience of dosing schedule, or rates of limiting side effects.
Moving forward, several unresolved questions remain in the field of PAH. Firstly, none of the currently available therapies is curative, so the search for new treatment strategies continues. As we increase our understanding of specific disease pathways involved with PAH, there remains the possibility of developing targeted therapies that will further improve outcomes. Secondly, there remains a need to identify patients with PAH earlier, before the onset of extensive vascular remodeling. New tools and diagnostic techniques will be essential to deriving maximal benefit from our expanding therapeutic armamentarium.
Finally, to optimize the management of PAH patients, the central role of the clinical nurse and a multidisciplinary team should not be overlooked. These individuals, with expertise in case management, collaboration, process improvement, and clinical skills, ensure adherence to therapy in addition to improving the quality of life of patients with PAH. Accordingly, clinicians should be encouraged to foster the development of nurse-led multidisciplinary teams and specialized PAH centers.
Disclosure
This manuscript has been read and approved by all authors. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The authors and peer reviewers of this paper report no conflicts of interest. The authors confirm that they have permission to reproduce any copyrighted material.