
Abstract
Pulmonary arterial hypertension (PAH) is a deadly and underdiagnosed disease which causes right heart failure secondary to pressure overload resulting from the thickening of the pulmonary artery endothelium, associated with elevated levels of circulating endothelin-1. Sitaxentan was the first endothelin antagonist with high selectivity for receptor subtype A (over subtype ET-B) to gain regulatory approval for the treatment of PAH in major pharmaceutical markets. This review traces the development history of sitaxentan, summarizes the designs and results from its clinical studies, and relates the drug's profile to that of the two other broadly available endothelin receptor antagonists, bosentan and ambrisentan. All three drugs have comparable therapeutic efficacy in the 6-minute walk test–-a frequently employed standard–-during the first 3-4 months of therapy. Their performance might differ slightly in other clinically relevant outcome measures, especially in longer-term treatment where fully comparable data have not yet been reported. In clinical trials of up to one year duration the propensity of sitaxentan to induce elevation of liver transaminases and hepatic failure was significantly lower than that of bosentan. As a once-daily oral drug with good tolerability sitaxentan has become a crucial element in the treatment of PAH.
Pulmonary Arterial Hypertension: An Endothelium-Related Disease
Pulmonary arterial hypertension (PAH), defined as a persistent mean pulmonary filling pressure of >25 mmHg at rest or >30 mmHg during exercise with normal “left-sided” arterial pressure, results from a narrowing of the lumen of the pulmonary artery. The initial symptoms–-shortness of breath and dizziness which are exacerbated by minimal exertion–-are characteristic but frequently misinterpreted as a simple lack of physical stamina. If patients seek medical attention they are correctly diagnosed only with considerable delay, in many cases not until they present to a cardiorespiratory specialist. As a result, the majority of PAH patients have already progressed to World Health Organization functional class III (characterized by a marked limitation of everyday physical activities) before a correct diagnosis is made, and targeted treatment is initiated. Eventually, the chronic pressure overload of the right ventricle results in right heart failure.
The main factors believed to contribute to the pulmonal arterial pathology are vasoconstriction caused by a relative deficiency of vasodilatory factors, and a proliferation of the arterial endothelium. Endothelin-1, a highly vasoconstrictive peptide released by the endothelium, is a key factor in the pathogenesis. 1 PAH as a primary disease is mainly idiopathic and although several genetic factors are known that confer elevated familial risk, the ultimate cause remains unclear in most cases. Idiopathic PAH is a rare disorder (incidence, 2-3 cases per million individuals and year; prevalence, approx. 15 cases per million) that must not be confused with the much more prevalent “simple” pulmonary hypertension which develops secondary to left heart disease, chronic pulmonary disease, or acutely in response to reduced oxygen partial pressure at high altitudes.
PAH developing in the context of systemic connective tissue diseases (where endothelin-1 also plays a crucial role in pathogenesis) represents a distinct etiology. It is more frequent than the idiopathic form and tends to be less responsive to treatment. The thickening of the pulmonary artery endothelium can also develop as a side effect of vessel malformations, thrombotic disorders, infection with the human immunodeficiency virus, or liver disease, and has been associated with amphetamine anorectic medications such as phentermine. 2
Drugs and drug candidates that are available to treat PAH or are in advanced stages of development have recently been reviewed.3,4 The currently most prominent class, suitable for the oral treatment of all but the most severe cases (patients in functional class IV are to be initiated on prostacyclins) or the neonatal forms of PAH (mostly treated with nitric oxide), are the endothelin receptor antagonists (ERAs) which block the proliferative and fibrotic effects of the endothelins. The prototypic ERA, bosentan, inhibits both endothelin receptor subtypes (A and B) that are present in the pulmonary artery endothelium. Because ET-B receptors continue to mediate vasodilation, vasoprotection and endothelin-1 clearance from the bloodstream despite the pathology associated with pulmonary hypertension, 5 and because their endothelin-mediated activation in the kidney is important for renal blood flow and natiuresis, it might be advisable to spare these potentially beneficial functions. ET-A receptor selective ERAs have therefore been developed to provide a more targeted therapy which attempts to block the deleterious vasoconstrictor and vasoproliferative effects of endothelin on vascular smooth muscle cells.
The first subtype-selective ERA to reach major markets, sitaxentan (ATC Code: C02KX03), is the subject of this review. (It should be noted that although the term “sitaxsentan” has become dominant in the literature, the approved international non-proprietary name (INN) of the molecule is actually sitaxentan, without a second “s”).
Discovery and Preclinical Characterization of Sitaxentan
In 1994 a United States patent application was filed 6 describing the synthesis and characterization of a series of 3-thiophenesulfonamide endothelin receptor antagonists. This series included a compound designated TBC-11251 which was later renamed sitaxentan (Fig. 1). (The research code refers to Texas Biotechnology Corporation, which later changed its name to Encysive Pharmaceuticals, and was taken over by Pfizer in 2008.) This molecule has considerable structural similarities with BMS-182874, an investigational ET-A receptor antagonist with high selectivity (created by Bristol-Myers Squibb, now discontinued from development) for which a receptor docking model was later proposed 7 which might have some validity for sitaxentan. Bosentan, the non-selective endothelin antagonist which entered clinical development at this time, is also a sulfonamide but otherwise its chemical structure is quite different from that of sitaxentan.
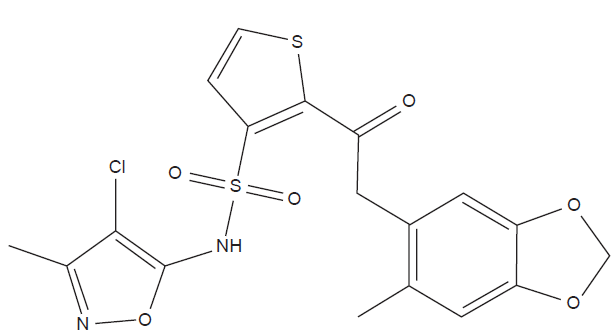
In the first peer-reviewed paper discussing TBC-11251, 8 the inventors reported competitive binding to human endothelin receptors with an IC50 value of 1.4 nM for ET-A receptors and an IC50 of 9800 nM for ETA-B receptors. Seen in conjunction with the favorable bioavailability and pharmacokinetic data in rats and dogs that were also presented, this high receptor subtype selectivity (7,000-fold in favor of the ET-A receptor) suggested that sitaxentan might hold exceptional promise for the treatment of PAH. A study performed at the Karolinska Institute (Stockholm, Sweden) had confirmed that TBC-11251 reduced the systemic vasoconstriction evoked in normoxic pigs by endothelin-1 infusion. 9 In rats, the pulmonary vasoconstrictor response to two weeks of chronic hypoxia (air oxygen content, 10% [v/v] instead of the normal 20%) and the associated right ventricular hypertrophy were attenuated, and remodeling of small pulmonary arteries was prevented, without affecting systemic arterial blood pressure or heart rate; a selective ET-B receptor antagonist was without effect. 10 Similar observations were made in horses.11,12
By this time published data obtained with other animal models had suggested additional therapeutic utility of TBC-11251 in the treatment of cerebral vasospasm 13 and retinal ischemia. 14
Studies with human hepatocytes and hepatic microsomal preparations have established that sitaxentan (and two of its metabolites which have been characterized) are inhibitors of liver cytochromes 2C9 and to a lesser extent, 2C19 and 3A4/5 (and possibly also of 2C8). The CYP2C9 blockade indicates a potential for drug interactions with vitamin K antagonist anticoagulants (such as warfarin), which can be relevant in the therapy and prevention of thrombosis in PAH. Further in vitro studies with transporter proteins demonstrated that sitaxentan is not a substrate for p-glycoprotein but might be a substrate for the human hepatic organic anion transporting polypeptide (OATBP) 1B1. 15
Clinical Studies
Phase I pharmacokinetic and drug interaction studies
Initial human pharmacokinetic information on sitaxentan was obtained using data from five Phase I studies (FNL01, FNL02, FNL06, FNL08A, and FNL13). These studies have not been reported in the peer-reviewed literature but summary results are available from the EPAR Scientific Report published by the EMEA. 15
The absolute bioavailability of sitaxentan sodium is 70%-100%. After administration of 25 and 100 mg coated tablets the median Tmax ranged from 0.5 to 4 hours, and a single 100 mg dose produced mean Cmax values of 7-13 μg/mL. At steady state conditions, which were established after 5 days of once-daily daily 100 mg dosing in fasting healthy individuals, the Cmax, Ctrough, and AUC0-24 values were 13 ± 5 μg/mL, 0.06 ± 0.07 μg/mL, and 40 ± 26 μg.h/mL, respectively (mean ± SD). Sitaxentan is almost completely (99%) bound to plasma proteins at these concentrations. The terminal half-life after single dose and steady state administration of 100 mg sitaxentan was approximately 7 hours. A standard meal decreased Cmax by 43% and doubled Tmax but did not influence the total systemic exposure; therefore no dose adjustments are required if sitaxentan is taken with food.
To follow up on the in vitro data concerning cytochrome metabolism inhibition, a drug interaction study (FNL02) was conducted. With 100 mg once daily doses of sitaxentan the AUC0-∞ of 25 mg S-warfarin (a CYP2C9 substrate) increased 95% and the terminal half-life of warfarin was increased, however, without significantly changing the Cmax of S-warfarin. In patients anticoagulated with a stable warfarin regimen this interaction requires a reduction of the warfarin dose when sitaxentan is initiated and patients need to be checked for their international normalized ratio (INR) coagulation status. No clinically significant interactions on the pharmacokinetics of nifedipine (CYP3A4/5 substrate), sildenafil (CYP3A4/5 substrate), ciclosporin (CYP3A4/5 substrate), omeprazole (CYP2C19 substrate), ketoconazole and nelfinavir (CYP2C19 and CYP3A4/5 substrates), or digoxin (p-glycoprotein substrate) were found after combined administration with sitaxentan.
Any two-way interaction with sildenafil might be considered particularly problematic because this drug is specifically approved for the oral treatment of PAH and, since its mechanism is different (phosphodiesterase-5 inhibiton) its combined use with an ERA seems very attractive. (Bosentan is known for such an interaction; at therapeutic doses it decreases sildenafil plasma concentrations by 55%, and sildenafil increases bosentan levels by 50%.) The part of study FNL02 that had investigated these interactions has been reported separately and in detail. 16 In a 24-patient crossover study, doses of 100 mg sildenafil did not change the plasma levels of sitaxentan given in 100 mg doses. Conversely, sitaxentan slightly increased the Cmax (by 18%) and the AUC (by 28%) of sildenafil but had no effect on its primary active metabolite, N-desmethylsildenafil. Systolic and diastolic blood pressure were comparable after administration of sildenafil with sitaxentan or placebo, and a similar percent of subjects reported adverse events when sitaxentan or placebo were added to sildenafil (39% and 30%, resp.).
Renal impairment, which is frequent in PAH patients suffering from scleroderma and other connective tissue diseases, has no influence on the pharmacokinetics of sitaxentan. 17 No pharmacokinetic studies were conducted in patients with impaired hepatic function because ERA treatment is not recommended in such patients.
12-Week open-label phase II study (TBC11251-211)
In a first attempt at evaluating safety and efficacy of sitaxentan, 6 children and 14 adults with New York Heart Association (NYHA) functional class II, III, or IV primary pulmonary hypertension (n = 8) or PAH associated with either congenital systemic-to-pulmonary shunts (n = 10) or collagen vascular disease (n = 2) received oral doses of 100 to 500 mg bid (depending on body weight; 4-6 mg/kg) for 12 weeks.
In spite of high standard deviations (see Fig. 2) which can be attributed to the heterogenous population and variable doses, sitaxentan treatment resulted in significant improvement as assessed by the 6-min walk distance (6MWD; the standard test for symptom-limited exercise capacity in cardiopulmonary conditions, where the distance which a patient can walk unencouraged during 6.0 minutes on level and smooth ground is measured 18 ). Patients improved from a baseline value of 466 ± 132 m (mean ± SD) to 515 ± 141 m at endpoint (p = 0.006). There was a significant correlation between change in 6MWD from baseline to week 12 and the corresponding change in mixed venous oxygen saturation (p = 0.029) but not in other hemodynamic variables. Mean pulmonary artery pressure and pulmonary vascular resistance index also improved (from 63 ± 20 to 52 ± 22 mmHg, n = 17, p = 0.0002; and 20 ± 11 to 14 ± 13 U × m2, n = 17, p = 0.008, respectively).
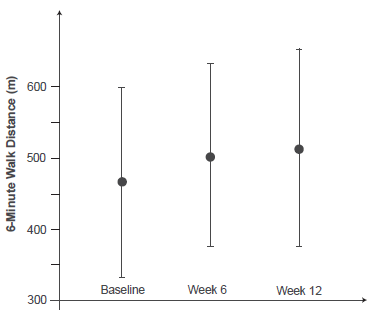
6-Minute Walk Distance at baseline and after 6 and 12 weeks of sitaxentan treatment in study TBC11251-211. Baseline and week 6, n = 20; week 12, n = 17 with inputed values for the three discontinued patients. 19
Mild, asymptomatic elevations in liver serum transaminase values from baseline to week 12 were observed in seven patients. However, two of these patients who had been enrolled in an extension study following completion of the 12-week trial developed acute severe hepatitis after a total of 16-17 weeks of sitaxentan treatment. While one patient's clinical and laboratory course normalized with a return to baseline serum liver function enzyme values approximately 9 weeks after discontinuation of the drug, the other one died. After the two patients had developed hepatitis in the extension period, all remaining patients were discontinued from the study drug, and safely tolerated discontinuation without clinically apparent episodes of rebound pulmonary hypertension. 19
Pivotal phase III trials
STRIDE-1 and its extension
The dose selection for the first pivotal clinical study was based on a combination of the experience from study TBC11251-211 and Michaelis-Menten modeling of pharmacokinetic data from normal human volunteers which had predicted that sitaxentan 100 and 300 mg qd should yield steady-state trough plasma free-drug concentrations of 1.4-fold and 6.1-fold in excess over the IC50 value, assuming 99.9% protein binding of the drug. Therefore, this dosing regimen was chosen in an attempt to reduce risk while maintaining efficacy. This Phase III trial was originally named FPH-01, but became publicly known as STRIDE-1 (Sitaxentan To Relieve ImpaireD Exercise).
The design of the double-blind, placebo-controlled STRIDE-1 trial was innovative but non-standard in two respects. While it is customary for pivotal PAH trials to enroll only advanced (WHO class III/IV) patients with idiopathic disease or PAH related to connective tissue disease with baseline 6MWD values of less than 450 m, STRIDE-1 was much more liberal: it included mild (class II) cases, allowed congenital heart disease, and employed no baseline 6MWD cut-off. More importantly even, it demoted the classical clinical endpoint accepted by regulatory authorities–-the change of 6MWD versus baseline–-to one of several secondary outcome measures while choosing percent of predicted peak exercise oxygen consumption (peak VO2; a physiological measure of exercise potential) as the sole primary endpoint. Peak VO2 is regarded as a good predictor of survival in PAH but is not tightly correlated with the 6MWD.
A total of 178 patients were enrolled between July 2001 and May 2002 at 23 centers (22 in the United States, 1 in Canada). PAH was idiopathic in 94 patients (53%), related to connective tissue disease in 42 (24%), and related to congenital vascular malformations (systemic-to-pulmonary shunts) in 42 (24%) patients. Sixty patients received placebo and 118 received sitaxentan (55 patients, 100 mg; 63 patients, 300 mg) for 12 weeks. At endpoint, only the 300-mg group improved its predicted peak VO2 compared with placebo (3.1%; p < 0.01); no improvement occurred in the 100-mg group. However, both groups increased their 6MWD almost equally by a mean of 20-22 m while the placebo group deteriorated by 13 m, i.e. the treatment effects versus placebo were 35 m (p < 0.01) for the 100-mg dose and 33 m (p < 0.01) for the 300-mg dose. Both doses of sitaxentan improved pulmonary vascular resistance (p < 0.001 for both doses) and cardiac index (p = 0.013 for 100 mg and p < 0.001 for 300 mg) compared with placebo. 20
This result was obviously not satisfactory: the Phase II pilot study had suggested that the 300 mg/day dose might not be sufficiently safe in the intended target group of patients for longer treatment (although STRIDE-1 had shown no difference in safety between the 100 mg and 300 mg doses during 12 weeks), and the 100 mg/day dose had failed the designated primary endpoint. Also, the results could not be easily related to those obtained in pivotal trials of approved PAH drugs (including the single approved ERA, bosentan) because of the nonstandard enrollment criteria and the choice of endpoints.
An intent-to-treat subgroup analysis of the STRIDE-1 data that focused on the 70 patients who would have met the classical inclusion criteria for PAH studies and on the 6MWD results was published a few months after the original results had entered the literature. 21 Because the 6MWD results for both doses of sitaxentan had been essentially equal in the STRIDE-1 intent-to-treat cohorts receiving active drug, the 47 “classical” patients on active drug were pooled into one treatment group for the purpose of this analysis, and compared to the 23 “classical” patients receiving placebo. (These subgroups remained sufficiently matched for their baseline clinical and demographic parameters to warrant this decision).
In this analysis, patients receiving placebo had a mean decrease in 6-minute walk distance of 26 m over the 12 week period of the study, while patients on sitaxentan showed an improvement of 39 m (P = 0.0002; placebo-adjusted treatment effect for sitaxentan therapy, 65 m). Therefore, the baseline-to-endpoint drug effect on the 6MWD in the patient group meeting the traditional enrollment criteria was almost twice as strong as in the more liberally enrolled entire STRIDE-1 cohort, and exceeded the treatment effect of bosentan in its 16-week BREATHE-1 trial. 22 Right atrial pressure (the key predictor of right ventricular dysfunction and disease progression in PAH) responded to the subset analysis in a similar fashion (mean placebo-adjusted treatment effect for sitaxentan therapy, -3.3 mmHg). In the following year the entire STRIDE-1 Study Group published a very similar analysis yielding almost identical results. 23
Another post-hoc subgroup analysis focused on the 42 STRIDE-1 patients with connective tissue disease,24,25 a subgroup which typically is more difficult to manage clinically than patients with idiopathic PAH and which has an inferior prognosis. Here the 6MWD treatment effect was 58 m (p = 0.0274), due both to an increase in 6MWD in the sitaxentan group from baseline (+20 m; p = 0.0327) and a decrease in 6MWD in the placebo group from baseline (-38 m). Improvements were largely preserved during the blinded extension phase of the study (see below); more patients were in functional class I–II than in III–IV (p < 0.001) at the end of the study compared with the start of active therapy. Elevation of hepatic transaminase levels occurred in two patients. A smaller (n = 23) and entirely independent Brazilian open-label study in patients with collagen vascular disease also found an 18 m 6MWD improvement versus baseline after 16 weeks, and a significant improvement in the physical component of the SF-36 quality of life score. 26
Upon completion of the 12-week STRIDE-1 study, 170 patients had entered a blinded extension (STRIDE-1X) and were treated with either 100 mg (n = 79) or 300 mg (n = 91) sitaxentan per day. Median treatment time was 26 weeks with exposure continuing up to 55 weeks. An additional 29% of patients on 100 mg and 15% on 300 mg improved their PAH functional class during weeks 12 to 24 with the remainder improving during weeks 26 to 55. During treatment, 5% of patients deteriorated on 100 mg and 8% on 300 mg. From baseline to the individual end of treatment, 42 patients on 100 mg (53%) and 40 patients on 300 mg (44%) improved at least one functional class. During the initial 12 weeks, liver function abnormalities (enzyme levels >3 × upper limit of normal [ULN]) occurred in 0% for 100 mg and 10% for 300 mg, with overall rates of 5% for 100 mg and 21% for 300 mg reported during the entire treatment course. 27
This evaluation had not reported results of exercise or hemodynamic testing, but another report on 11 Canadian patients, 28 10 of whom were treated with 100 mg or 300 mg sitaxentan for a full year, reported a 50 m improvement in the 6MWD, a sustained improvement comparable to what was achieved by bosentan in a similar long-term study. 29 No liver function abnormalities or serious adverse events were reported, and no clinically manifest interactions with warfarin were observed.
A further observational compassionate-use extension study, Stride-1XC, continued to treat 11 patients with sitaxentan for a total of 2 years. At the end of year 2, the remaining 9 patients were all in WHO functional class II and their mean 6MWD had improved from 387 ± 122 (SD) meters at baseline, to 436 ± 82 m at 1 year (p = 0.04), and to 440 ± 86 m at two years of sitaxsentan therapy (p = 0.02 versus baseline). 30
STRIDE-2 and its extension
For the next pivotal study, STRIDE-2, 31 the planners resigned themselves to a more classical design. In this double-blind, placebo-controlled 18-week study, 245 PAH patients (idiopathic, or associated with connective tissue disease or congenital heart disease; WHO functional class II–IV; baseline 6MWD 150-450 m) were randomized at 55 clinical sites. Based on the results of STRIDE-1 which had established the 100 mg dose as equally effective to, but safer than, the 300 mg dose, STRIDE-2 was powered to show superiority of 100 mg/day (to which 61 patients were randomized) over placebo (n = 62). However, a 50 mg/day sitaxsentan arm (n = 62) was also included, exploring clinical efficacy in a low-dose range that had not been evaluated before. Most notably, the study also comprised an observational open-label, rater-blinded arm of patients treated with bosentan (n = 60). Dosing in this observational arm adhered to the approved dosing regimen of bosentan, i.e. the drug was initiated at 62.5 mg twice daily for four weeks, then increasing to the maintenance dose of 125 mg orally twice daily. In contrast to prior long-term clinical PAH trials, the addition of any long-term PAH therapy was not allowed in STRIDE-2 in order to maintain the best possible comparability between the two active drugs.
Patients receiving sitaxentan were discontinued from therapy if they had an elevation in alanine aminotransferase (ALT) or aspartate aminotransferase (AST) levels > 5 × ULN, or if they had an elevation in total bilirubin > 2 × ULN with ALT and/or AST > 3 × ULN. For patients receiving bosentan, liver function abnormalities were handled according to the drug's approved product insert, which specified more complicated discontinuation criteria with somewhat higher limits for liver function parameters. The primary end point was change in 6MWD from baseline to week 18. Secondary end points included change in WHO class, time to clinical worsening, and change in Borg dyspnea score.
Comparison of baseline characteristics and results in the entire intention-to-treat (ITT) population and in the “traditional enrollment criteria” complement of STRIDE-1 patients (Data Source). 23
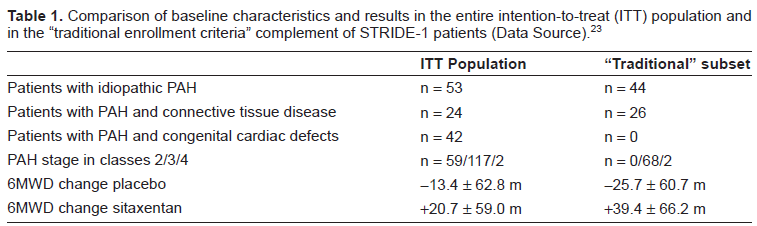
For the 100 mg/day sitaxentan dose, the results were similar to those of STRIDE-1: the 6MWD improved by a mean of 31.4 m versus placebo (p = 0.03), essentially identical to the 29.5 m (p = 0.05) treatment effect in the bosentan comparison group (see Fig. 3). The 50 mg/day sitaxentan regimen did not reach this performance, and was not statistically significant (6MWD improvement 24.2 m, p = 0.07). The incidence of elevated hepatic transaminases (>3 × ULN) was 6% for placebo, 5% for sitaxentan 50 mg, 3% for sitaxentan 100 mg, and 11% for bosentan; these incidences closely approximated what had been expected from the literature. Ninety-eight percent of the sitaxentan 100-mg patients improved their WHO functional class (13%) or remained unchanged (85%), whereas 87% of the placebo patients improved functional class (10%) or remained unchanged (77%). No significant improvements were seen in this parameter after 18 weeks versus placebo in either the sitaxentan 50 mg group or the bosentan group.
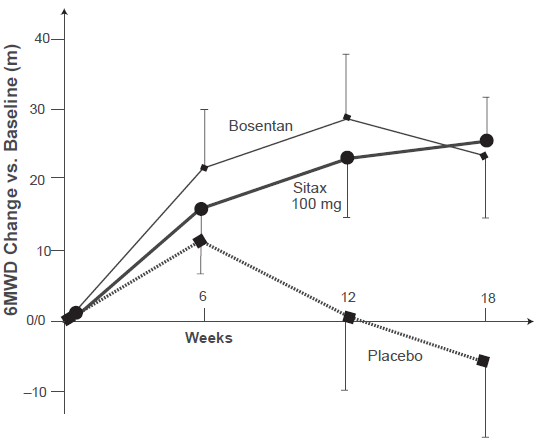
Mean changes (±SD) vs. group baseline in the 6-minute walk distances for placebo, sitaxentan 100 mg, and bosentan in the STRIDE-2 study. 31
STRIDE-2 also had an extension phase, STRIDE-2X into which patients were eligible to enter if they did not fail full-dose sitaxsentan or bosentan, or if they had prematurely discontinued placebo or sitaxsentan at 50 mg in STRIDE-2 due to clinical worsening. Eight patients who had received placebo and nine patients who had received sitaxentan 50 mg in STRIDE-2 did not continue in the extension and were excluded from all extended-duration analyses. Continuing patients who had received placebo in STRIDE-2 were randomly assigned in a 1:1 ratio to receive either sitaxentan 100 mg (n = 31) or bosentan (n = 23) for total exposure timed up to 52 weeks. The overall population enrolled in STRIDE-2X included 145 patients receiving sitaxentan at 100 mg qd and 84 patients receiving bosentan at 125 mg bid. Approximately 60 percent had idiopathic PAH, 30% had PAH due to connective tissue disease, and 10% had PAH secondary to congenital heart defects. For patients on active drug, the individual STRIDE-2 baseline values were used; for STRIDE-2 placebo patients, new baseline values were recorded upon randomization to active drug.
Following presentation of interim data,32,33 full one-year results of STRIDE-2/2X were published in late 2008. 34 Although this was not a full-scale double-blind comparison between sitaxentan and bosentan, it was the first head-to-head study with two ERAs in a clinical setting and correspondingly it attracted considerable interest. The analysis did not evaluate exercise parameters but focused on clinical progression of PAH, survival, and side effects of drug therapy (see Table 2).
Summary of STRIDE-2/2X results at the 1-year endpoint, by active drug. 34
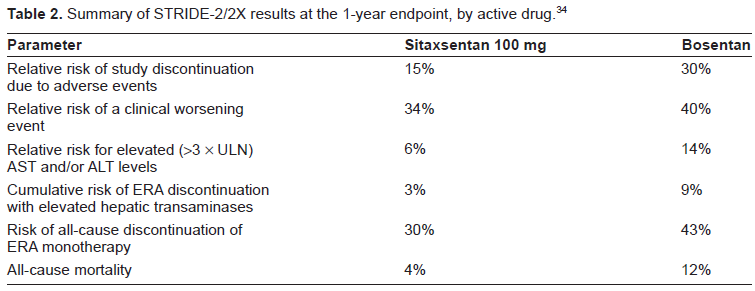
Kaplan-Meier analysis showed 70% of all sitaxentan-treated patients and 57% of bosentan-treated patients remaining on ERA monotherapy at 1 year, a discontinuation rate that was higher than expected from previous reports. A subgroup analysis indicated that these figures were 24% for sitaxsentan and 57% for bosentan in the 52 patients with connective tissue disease, in contrast with 32% and 37% in the remaining patients with PAH from other causes. Due to the design of the study, the post-hoc nature of the analysis, and the relatively small patient numbers it cannot be concluded yet that this is due to a true differential response.
Sitaxentan for patients discontinuing bosentan
A double-blind (but not placebo-controlled) study named STRIDE-6 randomized 48 patients who had discontinued bosentan therapy because of safety concerns (n = 13; 12 because of treatment-emergent hepatotoxicity) or inadequate efficacy (n = 35) to a single daily dose of either 50 mg or 100 mg sitaxentan. With 100 mg sitaxentan, 5 of 15 patients (33%) who discontinued bosentan because inadequate efficacy improved, demonstrating a > 15% increase in 6MWD, vs 2 of 20 patients (10%) treated with 50 mg sitaxentan. Fifteen percent and 20% of these patients had a >15% increase in 6MWD in the 50- and 100-mg groups, respectively. Similar results were seen for the Borg dyspnea score and the WHO functional class. Of the 12 patients discontinuing bosentan because of hepatotoxicity, 1 developed liver enzyme elevation at 13 weeks of sitaxentan therapy. 35
The SR-PAAS program: Combination with sildenafil
Unlike bosentan (and similar to ambrisentan), sitaxentan shows no potentially problematic pharmacokinetic interaction with the phosphodiesterase-5 inhibitor, sildenafil. During the second half of 2008 Pfizer began organizing a Phase III clinical study program to investigate the combined use of sitaxentan (which it had acquired through its takeover of Encysive Pharmaceuticals) with Pfizer's own approved formulation of sildenafil for the treatment of PAH. This ongoing program, named Sitaxentan Efficacy and Safety Trial With a Randomized Prospective Assessment of Adding Sildenafil (SR-PAAS), consists of three studies conducted in the United States.
The first study (ClinicalTrials.gov identifier NCT00795639) constitutes the introductory phase to the program and at the same time it is intended to provide the additional data which the U.S. Food and Drug Administration has required for the regulatory approval of sitaxentan as a monotherapy. It commenced patient enrollment in November 2008 and investigates sitaxentan (100 mg/day for 12 weeks) versus placebo in a planned total of 180 patients with Class III PAH, primarily for change in the 6MWD. Conclusion is anticipated for June 2010.
The second trial (NCT00796666) started in January 2009 and is open to patients who have completed the first trial. It adds sildenafil (3 × 20 mg/day) or sildenafil-matched placebo to sitaxentan monotherapy for a total of 48 weeks, with the final data collection date for the primary outcome measure (time to clinical worsening of PAH) planned for June 2011.
The third trial, NCT00796510, is an open-label safety study during which patients who either completed or discontinued the first trial protocol will receive either 100 mg sitaxentan or 3 × 20 mg sildenafil for 24 weeks. This study commenced patient recruitment in March 2009, with an envisaged completion date in January 2014.
Comparison of sitaxentan data with those of other ERAs
There are currently three ERAs approved for the treatment of PAH in major regions of the world: the non-selective antagonist, bosentan; and the two ET(A) subtype-selective drugs, sitaxentan and ambrisentan. How does sitaxentan perform with respect to its two competitors?
In terms of 6MWD after 12 weeks of doubleblind treatment the newest ERA, ambrisentan, achieved a maximal mean improvement of 59 m relative to placebo at the 10 mg/day dose which is not universally tolerated; 36 while the ceiling effect for the 100 mg/day sitaxentan standard dose was 65 m in the “population normalization re-analysis” which put the STRIDE-1 data on a comparable basis. (It had been 35 m in the full study, which had included patients in disease stages and with characteristics which trials with other drugs excluded.) For bosentan in its BREATHE-1 study, the placebo-corrected 16-week treatment effect on 6MWD was 35 m for 125 mg/day and 54 m for 250 mg/day, 22 a dose which is sometimes not tolerated. By just looking at the 6MWD group means it is evident that these three drugs achieve equivalent improvements in this key parameter, especially if it is considered that variances between individual patients tend to be large and that the improvement typically amounts only to 10%-15% of the baseline value.
Other clinically relevant endpoints such as time to clinical worsening and WHO functional class seem not to be improved as consistently by all agents, 37 however, these are not continuous variables and comparing these outcomes across studies is fraught with many caveats. In STRIDE-2 which allowed a first direct comparison (though not a rigid one) between sitaxentan and bosentan, sitaxentan 100 mg (double-blind) showed a trend towards prolonging the time to clinical worsening versus placebo while bosentan (open-label) did not. A similar trend was also seen in STRIDE-2 and its extension (see Table 2).
Hepatotoxicity has been referred to as a “class effect” of ERAs because all drugs with this type of activity that have been evaluated for the effect show it. However, not all ERA class members are equally affected. According to the basis of regulatory approval for the respective drugs, bosentan caused elevations (≥3 × ULN) of liver aminotransferases in about 11% of patients in the pivotal studies of 12-16 weeks duration. For sitaxentan this figure was 7% (against a background of 5% in placebo-treated patients), and it was only 0.8% for ambrisentan. However, longer-term observational studies, and the postmarketing surveillance programs to which all ERAs are subjected, indicate that incidence and prevalence of this side effect rises with the treatment duration, and might even manifest only after a year of constant treatment. There is still insufficient published evidence on this timescale to make a serious comparison between the three marketed ERAs. The fact that patients who had to be discontinued from treatment with one ERA can be safely re-initiated on another ERA35,38 indicates only that the mechanisms of liver toxicity might not be the same for these drugs.
There is not very much to be found in the literature that addresses the question whether elevation of liver transaminases is actually a direct effect of endothelin receptor inhibition. The hepatic endothelin system is not responsible for the increase of plasma endothelin-1 concentrations in acute liver failure, 39 and the effects of ERAs on cultured liver cells point more towards beneficial effects that counteract those of elevated endothelin levels. 40 It is possible that longer-term blockade of endothelin receptors in the hepatic vasculature interferes with angiogenesis (and therefor, with tissue turnover) in liver regeneration; the beneficial anti-angiogenetic effects of ERAs in solid tumors are well documented, as is their teratogenic potential. Shorter-term aspects of liver toxicity might be mediated by mechanisms of classical toxicology.
The pharmacoeconomics of sitaxentan seem comparable to those of the other two ERAs. For instance, the Scottish health authorities list the annual cost of sitaxentan therapy at GBP 20,020 and those for bosentan at GBP 20,033. 41
Summary
Sitaxentan, which is currently approved for the treatment of pulmonary hypertension in Europe and in several other countries (but not yet in the United States), is a selective endothelin A receptor antagonist which seems to have therapeutic efficacy similar to bosentan and ambrisentan. Its propensity for inducing liver damage is lower than that of bosentan but probably higher than that of ambrisentan; however, there are insufficient data available in the public domain to compare the time course characteristics of the transaminase elevation and actual clinical liver damage that occurs during longer treatment courses with these three drugs. Although the pharmacological rationale in favor of ERAs with high subtype selectivity for endothelin-A receptors is based on sound reasoning, it is not yet known if and to what extent this in vitro property translates to superior clinical efficacy and/or improved safety. Many other drug characteristics, e.g. tissue selectivity and pharmacodynamics, could diminish or enhance such characteristics in PAH patients.
Although the data on the performance of sitaxentan in the subgroup of PAH patients with systemic connective tissue diseases seems promising in terms of both efficacy and safety, 42 prospective clinical trials would be required to establish superiority over other PAH drugs.
With its generally good tolerability and its once-daily administration without the requirement for dose titration, the second-generation ERA sitaxentan seems definitely preferable over bosentan to initiate treatment in ERA-naive PAH patients. However, there seem to be few compelling reasons to switch patients who tolerate bosentan well and respond to the drug, unless compliance with twice-daily administration is an issue or if the intention exists to add sildenafil to the therapeutic regimen. (However, the marked pharmacokinetic interaction between bosentan and sildenafil might not be clinically relevant; see Gruenig et al). 43 There are much fewer differentiating criteria in evidence with respect to ambrisentan, and it would be interesting to see how sitaxentan and ambrisentan fare in a head-to-head comparison trial of at least one year duration.
Disclosure
The author was a consultant to Encysive Pharmaceuticals Germany in 2007 and 2008.