
Abstract
Since the mid-1970s, membrane modules became available and plasma separation techniques have gained importance. Therapeutic apheresis (TA) has been successfully introduced in a variety of autoantibody-mediated diseases. In dermatology, TA is increasingly applied as a support treatment for severe and/or refractory autoimmune bullous diseases. All autoimmune disease are characterized by autoantibodies mediated against structural proteins of the skin and/or mucous membranes. Autoimmune blistering diseases have a high morbidity and mortality. These diseases include pemphigus vulgaris and bullous pemphigoid among others. Drug-induced pemphigus and other forms, such as dermatitis herpetiformis, herpes gestationis, scleroderma, pyoderma gangrenosum, epidermal necrolysis, Behcet disease, psoriasis vulgaris, Henoch-Schönlein purpura, and porphyria cutanea tarda, are also mentioned. Pathogenetical aspects of autoantibodies in these diseases are clearly demonstrated. TA has been shown to effectively remove the autoantibodies from blood and lead to rapid clinical improvement.
Keywords
Introduction
The process of curing a patient by removing his illness by extracting blood is a very old one. Many years ago, phlebotomy was practiced to cure illness. Now, this old process, placed on a rational basis with plasmapheresis, is being followed in clinical practice. Until the development of hollow fiber membranes, therapeutic plasma exchange (TPE) was almost exclusively carried out by the centrifugal technique. Apheresis techniques have experienced a tremendous revival in recent years, and new therapeutic strategies have been established.
Therapeutic apheresis (TA) has been successfully used in various antibody-mediated diseases, such systemic lupus erythematosus, myasthenia gravis, Guillain-Barré syndrome, dilated cardiopathy, rheumatoid arthritis, idiopathic throm-bocytopenic purpura, hemophilia with inhibitors, dermatomyositis, ABO-incompatible kidney transplantation, and autoimmune bullous disorders. 1 The available adsorbers of more selective plasma separation methods can remove the nonspecific immunoglobulins from the patient's blood. 2 Since the pathogenetic relevance of autoantibodies could be defined in various diseases, disease-specific adsorbers have been developed, for example, in dilated cardiomyopathy (β1-adrenergic receptors), in systemic lupus erythematosus (C1q), and in grouping ABO blood group antigens.2–5 Besides the selective removal of immunoglobulins from a patient's blood, the adsorbers also remove immune complexes. All selective separation methods, such as various immunoadsorption (IA) techniques, full-blood adsorption, or other methods, which are mentioned here, are still technically complicated and expensive. The manufactures should develop simpler and less costly technique.
Some dermatologic immune-mediated diseases respond to TA. Responses are generally short lived, and repetitive procedures are generally required.6,7 In this review, the indications of TA in autoimmune blistering diseases and atopic dermatitis are discussed.
Methods
The first successful application of the membrane plasma separation in humans was reported by Yamazaki et al, Ash et al, and Castino et al in 1976.8–10 The advantages of this method are a complete separation of the corpuscular components from the plasma and higher efficacy due to increased blood flow rate. Furthermore, cell damage – especially to thrombocytes – that occurs when membrane hollow fiber are used is less than that occurring when centrifugation is used for cell separation. 2 Since the introduction of hollow fiber modules in TPE, this therapy is mostly used in nephrology. Nephrologists undergo extensive training in the management of blood purification treatments, including vascular access, anticoagulation, volume management, and prescription for solute clearance. 11
The diseases for which the use of TA is discussed and the guidelines on the use of TA provided by the Apheresis Applications Committee (AAC) of the American Society for Apheresis (ASFA) have been discussed in previous studies.12,13 TA methods, such as TPE, and semi- or selective plasma exchange methods, such as selective adsorbers, IA, were discussed by Bambauer et al. 14 Adsorptive cytapheresis mentioned in previous studies is a therapeutic procedure in which blood from the patient is passed through a medical device that contains a column or filter that selectively adsorbs activated monocytes and granulocytes and the remaining part of the blood is returned to the patient. 13 Extracorporeal photopheresis (ECP) is a procedure in which buffy coat, separated from patient's blood, is treated extracorporeally with a photoactive compound (eg, psoralens) and exposed to ultraviolet A light and subsequently reinfused to the patient during the same treatment. 13 In this paper, the authors give an overview of dermatological diseases in which TA is used or is indicated.
Dermatological diseases
Dermatologic immune-mediated diseases represent a heterogeneous group of disorders associated with circulating autoantibodies against distinct adhesion molecules of the skin and/or mucosa. According to the level of split formation, the disorders can be classified as intraepidermal blistering pemphigus, such as pemphigus vulgaris (PV), pemphigus foliaceus, and paraneoplastic pemphigus, and as subepidermal blistering pemphigoid diseases, such as bullous pemphigoid (BP), pemphigoid gestations, and dermatitis herpetiformis. 15 The newly developed sensitive and specific assays for circulating autoantibodies in these dermatological diseases enable now the serological diagnosis in about 90% of the cases.
The incidence of autoimmune blistering skin diseases in Germany has doubled during in the last 10 years, to about 25 new cases per million humans per year, because of improved diagnostic techniques as well as the age of the population. 15 There are an estimated 2,000 new cases of autoimmune blistering skin diseases being diagnosed per year. The incidence of pemphigus in Europe is one to two cases per million humans per year, and 80% of pemphigus patients have PV. 16 BP is the most common type of subepidermal autoimmune blistering skin disease in Europe, with an incidence of about 13 cases per million humans per year. The other common types are mucous membrane pemphigoid and pemphigoid gestationis. 17
The standard of diagnostic testing for autoimmune blistering skin diseases is direct immunofluorescence (IF) microscopy to demonstrate the presence of tissue-bound autoantibodies and/or of C3 in patients’ skin or mucous membranes. 18 Direct IF microscopy of the patient's serum can be used as a screening test for identifying circulating antibodies. The diagnostic assessment of autoimmune blistering skin diseases can be expected to improve in the near future as new serological testing systems are developed that employ recombinant forms of the target antigens. But the treatments in use still need to be validated by prospective, controlled trials. 15 As an example for intradermal blistering pemphigus, PV is discussed in the following section.
Pemphigus vulgaris
PV is a severe, chronic disease of the skin and mucous membranes and has poor prognosis and acantholytic blisters and erosion and is characterized by the presence of antibodies against epidermal intercellular substances. PV is a classic example of autoantibody-induced immune dermatosis, which can be recurrent or relapsing. The specific IgG fraction of the pemphigus serum initiates acantholysis without a complement. It is surmised that enzymatically induced destruction with plasminogen activator and pemphigus acantholysis factor occurs after binding of the pemphigus antibody to the surface of the epidermal cell. 19
Both genders are equally affected with the mean age of onset in the sixth and seventh decade of life, and the patients present with skin lesions that occur typically as flaccid blisters. 13 The blisters can be located on the entire body surface as well as on the mucous membranes of the mouth. A large area of the skin can be affected at any given point leading to situations akin to severe burn. PV is characterized by the deposition of an autoantibody on the keratinocyte cell surface. This antibody is typically directed against a 130-kDa protein (desmoglein 3). Additional autoantibodies against desmoglein 1 have been detected. Histology reveals the presence of a suprabasilar intraepidermal split with acantholysis, and there are deposits of IgG and C3 on the corticokeratinocyte cell surface in the mid and lower or entire epidermis of perilesional skin or mucosa. The titers of IgG4 antikeratinocyte antibodies can be correlated with disease activity. 13
PV was associated with a high morbidity and mortality. Introduction of corticosteroids reduced the mortality rate from 70% to 100% to a mean of 30%. 13 However, long-term administration of high doses of corticosteroids can be associated with severe side effects. Other therapeutic options include dapsone, gold, and systemic antibodies. They are often used in combination with other immunosuppressant agents, such as azathioprine, methotrexate, and cyclophosphamide. Newer therapeutic modalities, such as TPE, ECP, mycophenolate mofetil, chlorambucil, dexamethasone-cyclophosphamide, IVIG therapy, anti-CD20 monoclonal antiboldy (rituximab), have also been investigated. 13
The rationale for using TPE in the treatment of PV is based on the presence of circulating pathogenic autoantibodies. TPE has been utilized in patients with severe symptoms who received high doses of conventional agents and/or had an aggressive and rapidly progressive disease. TPE was used in patients in all age groups (13–80 years old). The duration of disease prior to using TPE ranged between 1 month and 25 years. The goal of TPE was to reduce the level of autoantibodies with subsequent improvement in clinical symptoms. The decline in autoantibody titers, antikeratinocyte cell surface antibodies, and anti–desmoglein-3 correlated with clinical response in a number of patients. 13
The antiepidermal antibodies, which usually belong to the IgG category, can be easily eliminated with TPE. Many authors have reported successful clinical results of TPE in the treatment of PV.19–23 According to Roujeau et al, TPE is indicated in pemphigus only after immunosuppressive therapy with a dosage of more than 2 mg/kg body weight (BW) of a prednisolone equivalent has failed. The success rate of a combined therapy of immunosuppression with steroids and TPE is over 95%.22,24 This was reported in recent years.19,25 Euler et al synchronized the TPE with pulse cyclophosphamide post-TPE to decrease rebound antibody production. This approach resulted in long-term remission in a 48-year-old patient with previously poorly controlled disease.
26
Standard therapy for PV is based on a combined administration of high-dose glucocorticoids and immunosuppressive drugs. In patients with severe, life-threatening, or recalcitrant PV, stronger therapeutic options should be considered, such as
Hashimoto proposed in 2008 treatment strategies for PV in Japan. 28 These include TPE, steroid pulse therapy, IVIG, various immunosuppressive agents (such as azathioprine, cyclophosphamide, cyclosporin, mycophenolate mofetil, and mizoribine), and rituximab. When all these therapies are used, care should be taken regarding the severe infectious diseases, except for IVIG. In the most intractable cases, a combination of these adjuvant treatments may be used. 28
IA has been successfully applied in patients with severe atopic dermatitis and high total serum IgE levels. 15 In the last few years, various IA systems and immunosuppressive protocols have been used to reduce the circulating autoantibodies. With a single IA procedure, between 50% and 75% of the specific pemphigus anti–desmoglein IgG autoantibodies can be eliminated by different adsorbers. 29 No differences in IgG depletion rates between the protein A–based and the human antihuman Ig–based reusable systems have been shown by Matic et al. 30 For obtaining durable effects of rapid decrease in circulating autoantibodies and immune complexes, IA must be combined with immunosuppressant treatment.
To date, different authors have reported on the treatment of PV with IA and immunosuppressive therapy, which was in all treated cases successful with complete remission, clinical remissions, or partial remissions.29,31–35 IA was performed as an adjuvant therapy in combination with different immunosuppression protocols. The autoantigen and the pathogenic relevance of autoantibodies are well documented. An antigen-specific IA would be the best method to induce rapid clinical response in PV patients.36,37 The complication rates and side effects are low and comparable with those of the other extracorporeal circulations. Recently, a large phase III trial evaluated the efficacy and safety of IA in pemphigus patients, but this technique was limited only for severe cases of PV. 38
Thus, in recent years, ECP has also been used in patients with serious cases of pemphigus with considerable success.39,40 The clinical response in patients who underwent the treatment of ECP was observed after two to seven cycles (two daily procedures per cycle). The total number of cycles received varied from 2 to 48. The follow-up ranged between 4 and 48 months, and the disease was controlled in most patients. 12
TPE protocols used in PV vary widely and have been usually based on the observed clinical response after each treatment. More recent reports noted that one plasma volume exchange is preferable in patients who are resistant to conventional therapy. 12 The levels of autoantibody have been noted to rebound in the reported patient within 1–2 weeks after discontinuation of treatment, which necessitates continuation of immunosuppression. 12 The treated volume is 1–1.5 total plasma volume (TPV), and the replacement fluid is an albumin–electrolyte solution and/or plasma. The frequency is daily or every other day. In the guidelines for the use of TA by the AAC of the ASFA, PV has category III for TPE, IA, and ECP with a recommendation of grade 2B and 2C, respectively (Table 1).12,13
Guidelines on the use of TA in dermatology.
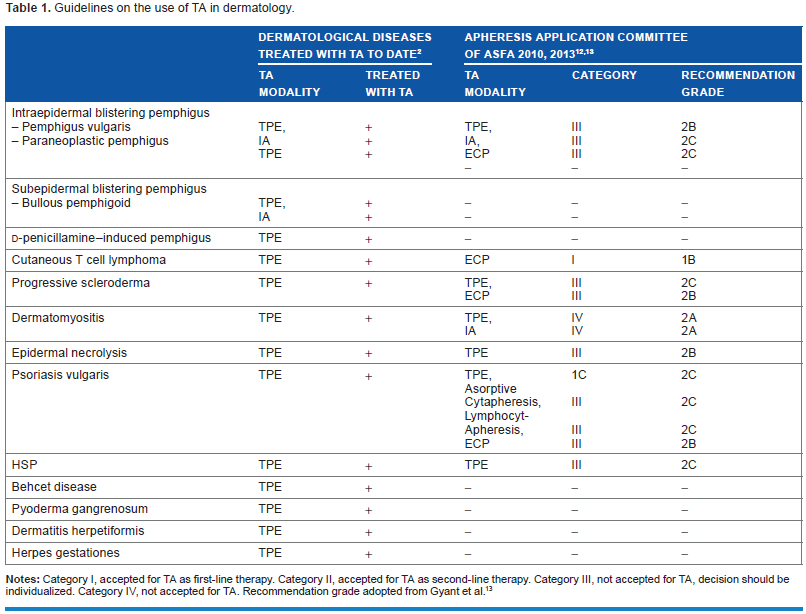
Notes: Category I, accepted for TA as first-line therapy. Category II, accepted for TA as second-line therapy. Category III, not accepted for TA, decision should be individualized. Category IV, not accepted for TA. Recommendation grade adopted from Gyant et al. 13
For TPE and IA, as noted above, the treatment protocols are highly variable. The rational approach should include monitoring of autoantibody titers and clinical symptoms. The lack of clinical response after a trial period with concomitant adequate immunosuppression should be sufficient to discontinue treatment. For ECP, the treatments were continued until clinical response was noted. The rational discontinuation criteria should be similar as those for TPE. 12
Bullous pemphigoid
Another form of subepidermal blistering pemphigus is the rare BP. BP frequently involves a premonitory stage with pruritic urticarial erythema and eczematous lesions followed by the classical bullous stage with tense blisters, erosions, and crusts. 15 BP is a chronic dermatosis often associated with acute exacerbations, with the formation of bullae blisters usually on the inflamed skin, subepidermal blister formation, and antibodies against the epidermal basal membrane. The pathophysiology is regarded as being a consequence of the combined effect of antigen, antibody, complement, and inflammatory cells, whereby lysosomal enzymes actually destroy the basal membrane zone and induce subepidermal blistering. 41 It is still unclear whether BP can be provoked by medication or ultraviolet rays. It is also possible that destruction of the basal membrane zone with release of basal membrane antigens can cause a direct immunological response in predisposed people. Thus, BP can also occur in combination with other autoimmune disorders. IgG autoantibodies against it are present in about 85% of patients with BP.42,43 Most BP patients also develop IgA and IgE antibodies against BP 180 (immunodominant region of BP 180), and 60%–70% of BP patients have circulating autoantibodies against BP-230,43–45 but the presence of these antibodies is much less specific than that of anti-BP-180 antibodies for the diagnosis of BP and is not correlated with disease activity. 44
However, because the clinical disease is not as potentially life threatening as other autoimmune diseases, the course of this pemphigus disorder is not as dramatic as other forms of the disease, with good response to high-potency corticosteroids, which are usually combined with dapsone, doxycycline, methotrexate, or azathioprine. 15 BP has an annual incidence of about 13–42 new cases per 1 million in central Europe and the United Kingdom.17,46 Only a few cases have been treated with TPE up to now.
The authors describe here a particularly severe case of BP that was favorably influenced by plasmapheresis. A 46-year-old female patient was first admitted to hospital as an inpatient with a severe bullous skin disorder of unclear etiology. 2 First, steroid as local treatment was tried but without success. As the BP disorder progressed rapidly, systemic corticoid therapy was applied for several weeks, also without satisfactory response and therapeutic effect. Upon further progression extending over the whole integument after 6 months of conservative therapy, TPE treatment commenced. Initially, some problems with vessel access were encountered; nevertheless, after only a few TPE sessions, there was a distinct improvement in skin efflorescence, with almost complete healing. Upon stopping TPE, the disease flared up again. TPE was restarted and remission was again achieved. The patient continued TPE intermittently for several weeks after achieving remission. This resulted in remission for 9 months. Again the flare was abated with TPE. Upon reduction of TPE treatment, the disorder flared up again, and the patient underwent intermittent TPE treatment. Another severe attack occurred after a fresh cell therapy had been administered to the patient. This new outbreak was brought under control with TPE. Occasional flares occurred over the next 4 years and each resolved with TPE. The patient was then lost to follow-up. In the following 2 years, there were two more outbreaks, each time after the patient had discontinued the therapy and had undergone treatment with fresh cells or similar treatment against the authors’ recommendations. The further course of the disorder after the 4-year treatment is unknown, as the patient did not continue therapy after the skin had almost fully healed. The poor prognosis for this disorder indicated intermittent TPE treatment, as the BP flared up each time the therapy was stopped.
IA has been successfully applied in patients with severe BP and can be performed with different single-use adsorbers or reusable adsorbers. The latter are much more effective than the former, enabling a 75% reduction of autoantibodies in a single IA and 95% reduction when IA is performed for 3 consecutive days.1,47 Various protocols for the use of IA in BP have been tested in combination with immunosuppression.1,29,42 In all studies, the introduction phase consisted of three or four IA treatments on consecutive days, usually with high-affinity adsorbers. All patients benefitted from the treatment. The main advantage of IA is its rapid clinical effect. It often results in the healing of all lesions within a few days.
Because the pathogenic relevance of autoantibodies was clearly demonstrated in the majority of autoimmune bullous diseases, removal of autoantibodies, therefore, appears to be a rational therapeutic approach for these patients. IA has been shown to effectively lower the antibody levels and leads to rapid clinical responses in patients with immunobullous disorders. 1 Meanwhile, IA and the monoclonal anti-CD20 antibody (rituximab) have been established as additional therapeutic options.47,48
D-Penicillamine–induced pemphigus
D-Penicillamine–induced steroid-resistant pemphigus (Table 1) is a foliaceus type disease with high lethality and mortality rate, which can occur as a side effect in long-term penicillamine therapy, which is a particular indication for TPE.49–51 The mechanism by which this drug induces acantholysis of the epidermis has still not been clarified. Most investigators suspect that immunological processes similar to those in PV are involved. The final step in anti-Dsg–induced acantholysis is the response of the keratinocyte to autoantibodies binding via downstream signaling events and eventual keratin filament retraction and apoptosis, as many signaling pathways have been implicated in anti-Dsg–induced acantholysis. 51
In drug-induced pemphigus, it was demonstrated that autoantibodies have the same antigenic specificity, on a molecular level, as autoantibodies from other pemphigus patients.52,53 The chance of acquiring pemphigus after penicillamine intake of least 6 months is 7%. Subsequently, more medications have been reported to evoke pemphigus, such as penicillin, ampicillin, rifampicin, pyrazolone derivatives, a combination of aspirin and indomethacin, and a combination of propanol and mepbromate.
54
Drugs
Bahmer et al presented data on a 45-year-old patient who had been treated for severe rheumatoid arthritis for over 2 years with a daily dose of 600 mg d-penicillamine. 57 Due to the severity of the pemphigus condition, the patient was admitted to the hospital for inpatient treatment. Despite high-dose corticosteroid therapy 100 mg daily over a period of 3–4 weeks, his condition kept deteriorating. Therefore, plasmapheresis treatment was commenced. After only a few sessions, the skin improved, and after approximately 15 sessions, the pemphigus was almost completely healed. A relapse 2 months later was also rapidly and positively influenced by further plasmapheresis treatment.
Immunosuppressive therapy is directed at preventing or slowing the rebound of antibody and immune complex formation after TA so as to sustain the therapeutic effect. Given that suppression of a rebound through renewed antibody synthesis is very important for the further course of the disease, however, studies by Roujeau et al show that cyclophosphamide in a daily dosage of 2–3 mg/kg BW is not sufficient to prevent the rebound effect. 58 In the following years, only case reports of d-penicillamine–induced pemphigus treated successfully with TA were reported. It is generally recommended to combine TA with immunosuppression. This time-consuming treatment has to be repeated in short time intervals.
IA is the most specific therapeutic option, in which only the pathogenic IgG is depleted in the patient's plasma. IgG autoantibodies are adsorbed on antihuman IgG affinity agarose column. Resynthesis of IgG autoantibodies was inhibited by postapheresis IVIG; therefore, the additional effect of IA is difficult to observe since IVIG has also a immunomodulatory potency. 15 A combination of IA and rituximab showed rapid and long-lasting response of concomitant immunosuppressive medication. 48 Rituximab is almost given as an adjuvant drug, ie, in addition to another type of immunosuppressive treatment. Complications of rituximab in patients with autoimmune blistering skin diseases include infections, deep venous thrombosis of the lower limbs, pulmonary embolism, long-term hypogammaglobulinemia, and neutropenia with an overall mortality of 4%. 1 The indications, contraindications, and dosage of rituximab treatment for autoimmune blistering skin diseases as well as the variables that should be monitored over the course of treatment and the criteria for discontinuing rituximab have been established. 59
Other forms of dermatosis
Myosis fungoides and its leukemic variant, Sézary syndrome (SS), are the most common types of cutaneous T-cell lymphoma (CTCL) whose pathogenesis remains elusive (Table 1). 12 CTCL is incurable. Therapy is aimed at alleviating symptoms, improving skin manifestations, controlling extracutaneous complications, and minimizing immunosuppression. 13 Chemotherapy is recommended for aggressive SS, with alemtuzumab and stem cell transplantation being considered for refractory disease.
In CTCL ECP is indicated. ECP involves the collection of circulating malignant CD4+ T cells, ex vivo treatment with 8-methoxypsoralen and UVA light and reinfusion of the cells. The therapeutic effect appears to be mediated by
ECP should be planned for a minimum of 6 months. When maximal response is achieved, it can be reduced to once every 6–12 weeks. If there is evidence of disease progression after 6 months of ECP alone, combination therapy should be considered. If there is minimal or no response after 3 months of combination therapy, ECP should be discontinued. 12
When combined with immunosuppression, TPE is probably successful due to the pathogenesis of severe cases of dermatitis herpetiformis and herpes gestationis.60–64 Herpes gestationis or pemphigoid gestationis is an autoimmune subepidermal blistering disease that occurs in women in the second or third trimesters of pregnancy or even puerperium. It is a rare skin disease, the incidence of which has been estimated of approximately one case in every 40,000–60,000 pregnancies. 65 In patients who are resistant to corticosteroids, other treatments have been tested including immunosuppressant drugs, such as cyclosporin, azathioprine, and tacrolimus and plasmapheresis. 65 In contrast, dermatitis herpetiformis produces anti–desmoglein-3 autoantibodies and could be a variant of PV with unique clinical and histological features. 66
Scleroderma or systemic sclerosis is a rare, generalized autoimmune disease. Scleroderma is characterized by vascular abnormalities, fibrosis, inflammatory changes, and late-stage atrophy/obliterative vasculopathy. Localized scleroderma forms show a longitudinal or circumscribed skin involvement. 65 The effectiveness of TPE in progressive scleroderma and dermatomyositis is still disputed.
Pyoderma gangrenosum (PG) is a rare, polyetiological syndrome based on a pathological immune reaction. 67 In over 40% of cases, this disease occurs together with colitis ulcerosa. In the vessel walls of vasculitic lesions, granular IgG, C3, complement, and IgM deposits have been observed. 68 PG is a noninfectious neutrophilic dermatosis that usually starts with sterile pustules that rapidly progress to painful ulcers of variable depth and size with undermined violaceous borders. In 17%–74% of cases, PG is associated with an underlying disease, most commonly inflammatory bowel disease, rheumatological or hematological disease, or malignancy. Diagnosis of PG is based on a history of underlying disease, typical clinical presentation and histopathology, and exclusion of other diseases that would lead to a similar appearance. 69 PG is characterized by painful, enlarging necrotic ulcers with bluish undermined borders surrounded by an advancing zone of erythema; its clinical variants include ulcerative or classic, pustular, bullous or typical, vegetative, peristomal, and drug-induced. Subcorneal pustular dermatosis is an uncommon relapsing symmetric pustular eruption that involves flexural and intertriginous areas; it can be idiopathic or associated with cancer, infections, medications, and systemic diseases.70,71 Because the incidence of PG is low, no prospective randomized controlled trials and only a few studies with case numbers of more than 15 patients have been published. To date, no guidelines for the treatment of PG have been established.72,73
Weber et al reported the treatment of a 38-year-old female patient with a manifestation of PG, together with colitis ulcerosa. 74 The skin lesions worsened even with high doses of methylprednisolone. Immediately after a TPE treatment, the healing process began. Based on the report by Beurey et al. 68 , the postulated immunopathogenesis, and the exacerbation of the condition with conventional therapy, plasmapheresis treatment was carried out. 73
Therapeutic efficacy of systemic treatment with corticosteroids and cyclosporin is documented in the literature for disseminated as well as for localized disease and should be considered as first-line therapy. In cases that do not respond to this treatment, alternative therapeutic procedures (eg, systemic corticosteroids and mycophenolate mofetil; mycophenolate mofetil and cyclosporin; tacrolimus; infliximab, or TPE) are recommended. 72 Despite recent advances in therapy, the prognosis of PG remains unpredictable.
Drug-induced toxic epidermal necrolysis (TEN), also known as Lyell's syndrome, is a life-threatening drug reaction characterized by extensive destruction of the epidermis and mucosal epithelia. The eyes are typically involved in TEN. The disease has a high mortality rate. TEN and the Stevens-Johnson syndrome (SJS) are closely related, although their severity and outcome are different. The SJS and TEN are rare but present severe skin manifestation. They are estimated to occur in one to three people per million per year in Europe and the United States. 75 They are characterized by a low incidence but high mortality, and drugs are most commonly implicated in 80% of TEN cases. 75 TEN is the most severe form of drug-induced skin reaction and is defined as epidermal detachment of >30% of total body surface area (TBSA). SJS presents with epidermal detachment of <10% of TBSA, whereas involvement of 10%–30% of TBSA is defined as SJS/TEN overlap. 76
Etiological, clinical, and histological characteristics help to distinguish TEN from severe forms of erythema multiforme. The current understanding of the pathomechanism of TEN suggests that keratinocytes are key initiator cells. It is probable that combined deleterious effects on keratinocyte of both are the cytokine. Tumor necrosis factor (TNF)-α and oxidative stress induce a combination of apoptotic and necrotic events. 77 There is an agreement to consider this phenomenon as the manifestation of a dysregulated immune reaction against epithelial cells. The systemic toxicity increases the risk for multiorgan failure. 78
In Lyell's syndrome, the acute phase can be very successfully treated by TPE. The allergic or toxin-induced skin necrolysis is usually triggered by a drug acting like a hapten. 79 Conleth et al reported in 1999 good results with TPE in the treatment of 16 patients with TEN. None of the TPE patients died. 80 According to Poulsen et al and Kostal et al, the immunological process can be promptly interrupted through implementation of TPE early in the course of the disease.81,82 Lyell's syndrome is fortunately very rare but has a high mortality rate, approximately 50%, and thus, early administration of TPE is justified. TPE is a safe intervention in severely ill TEN patients and may reduce the mortality in this severe disease.81,82 Other authors found that using IVIG in association with TPE may be effective in patients with severe TEN.83,84 The application of monoclonal antibody to TNF-α (infliximab) was also successful. 85 But prospective multicenter trials are needed to further define its usefulness.
Behcet disease, a multisystemic inflammatory disorder, presents with the involvement of muco-cutaneous, occular, vascular, central nervous, and gastrointestinal systems. It is an idiopathic, chronic, and recurrent disease characterized by exacerbation alternating with plasma of quiescence, episodic panuveitis, and aggressive nongranulomatous occlusive vasculitis of the arteries and veins of any size with explosive ocular inflammatory attacks that primarily affect the retinal and anterior segment vasculature of the eye. 86 Central nervous system involvement, most often due to necrotizing vasculitis, may be the most protean manifestation of the disease, leading to death. The frequency of ocular manifestations is 70%–85% in these patients. The underlying mechanism of this disease in all organ systems is an occlusive vasculitis. 87
It is more prevalent along the ancient Silk Route from the Mediterranean to East Asian countries, compared with Europe and America. Ethnic diversity can influence the progression severity and clinical manifestation.87,88 The effect of TPE in Behcet disease, where immunopathogenesis is also being debated”, is not clear. Although TPE has been successful in individual cases. 89 In recent years, there have been reports on the successful treatment with implementation of cyclosporin A, tacrolimus, or infliximab, etc.86,90,91
Psoriasis vulgaris is a common autoimmune chronic inflammatory skin disease that affects approximately 2% of the world's population. Fundamental for its immunopathogenic mechanism is the secretion of type 1 (Th 1) cytokines by T cells and their activation. 92 Cytokines are intercellular molecules that have an important role in the development and maintenance of cutaneous inflammation. 93 The indication for TPE in psoriasis vulgaris is also based on the presumption of immunopathogenesis. The pathogenesis of psoriasis is still for the most part unclear; however, autoantibodies, circulating immune complexes, and cytokines are thought to be responsible for triggering flairs of the disease or a new attack. 94 There is a correlation between antibody titer count and the clinical course of the disease.
In 1982, Beuthner et al, Rebora et al, and Valbonesi et al observed significant improvement in psoriasis with TPE.95–97 Valbonesi et al reported lasting improvement in seven out of eight psoriasis patients who had been treated with TPE. 97 On the other hand, Halevy et al were unable in 1982 to influence psoriatic erythroderma in an 18-year-old girl. 98 They only exchanged 500 mL in each of the 10 sessions, which was certainly not sufficient for any visible therapeutic effect. Jorstadt et al treated eight patients with psoriasis with skin scales and seven with disabling psoriatic arthritis with cascade filtration for 2 weeks. 99 There was a large drop in the levels of circulating immune complexes due to treatment, and the removal of circulating immune complex (CIC) was followed by reduced inflammatory activity in skin lesions and joints as evaluated by pain, morning stiffness, grip strength, plaque score, and Psoriasis Area Severity Index (PASI) index. However, there was no correlation between the level of CIC, disease activity, or treatment response. From these results, it is concluded that CIC may play a more significant role in psoriatic arthropathy than in skin manifestations, and TPE may be beneficial in patients not responding to conventional therapy. 99
Other authors reported their results with treatment of extracorporeal photochemotherapy and lymphocytapheresis. But the skin lesions did not respond to photopheresis.92,100
However, blocking TNF-α by infliximab or etanercept has shown particular promise, especially in the management of psoriasis. But further studies to address its mechanism of action and potential long-term side effects are needed.
Henoch-Schönlein purpura (HSP) is a systemic vasculitis that affects vessels of small size. The vascular purpura is usually confined to the lower limbs and is associated, at varying degrees, with joint, gastrointestinal, and renal involvement. 101 It is a systemic disease where antigen–antibody (IgA) complexes activate the alternate complement pathway, resulting in inflammation and small-vessel vasculitis. 102
In 1990, the American College of Rheumatology defined HSP as the presence of two or more of the following criteria: age of disease onset (20 years or younger), palpable purpura, acute abdominal pain, and granulocytic infiltration in the walls of arterioles or venules. 103 Focusing on the pathogenic role of IgA immune complexes in HSP, the Chapell Hill Consensus Group view the diagnosis as a small-vessel vasculitis with predominant IgA vascular deposits. All patients develop palpable purpura. In the skin, these deposits lead to subepidermal hemorrhage and small-vessel necrotizing vasculitis producing the purpura. 13 IgG autoantibodies directed at mesangial antigens may also play a role in pathogenesis. In other organs, necrotizing vasculitis leads to organ dysfunction or hemorrhage. Nonetheless, the precise role of IgA or antibodies in the pathogenesis of the disease remains unclear. 13
Patients with HSP were successfully treated with TPE by Camerone et al in 1982 104 and by Szpirt et al in 1984. 105 Other authors also reported successful treatment with TPE in patients with HSP.106–109
In the guidelines for the use of TPE by the AAC of the ASFA, the crescentic form and severe extrarenal manifestations of the HSP are classified as category III with the recommendation being grade 2C for TPE (Table 1). 13 Prospective randomized clinical studies proving treatment efficacy are still lacking. 110 Spontaneous recovery even in patients with severe clinical and histological presentation and of late evolution to chronic kidney disease in patients with mild initial symptoms renders it difficult for treatment protocols. Prospective international multicenter studies looking at determinants of clinical and histopathological evolution as well as possible circulating and urinary markers of progression are necessary. 111
Porphyria cutanea tarda (PCT), a genetic enzyme defect, was also considered by Distler et al in 1982 as being a treatable condition with possible indication for TPE. 112 PCT is a metabolic disorder of the hem biosynthesis caused by decreased activity of uroporphyrinogen decarboxylase. PCT is manifest by fragility, erosions, bullae, milia, and scars on sun-exposed skin. Excess porphyrins in the skin interact with light of approximately 400-nm–wave length radiant energy, forming reactive oxygen species. PCT is categorized as familial, acquired, or toxic. Factors that may induce clinical expression of PCT in susceptible individuals include alcohol, estrogen, iron, polyalogenated compounds, and viral infections. PCT is associated with an increased incidence of the hemochromatosis gene. 113
Recent advances in the pathogenesis are the identification of the iron overload–induced inhibitor of hepatic uroporphyrin decarboxylase activity that causes the most common porphyria, PCT, and the identification of an X-linked form of erythropoietic protophyria due to gain-of-function mutations in erythroid-specific 5-aminolevulinate synthase. 114
Protoporphyrin accumulates in the maturing red blood cells during hematopoiesis. When red blood cells enter the circulation, free protoporphyrin diffuses across the red blood cell membrane and binds to plasma proteins. The liver extracts protoporphyrin from the plasma, most of which is excreted unchanged into the bile, with the remainder being metabolized (by liver ferrochelatase) to hem. Some protoporphyrin is subsequently reabsorbed during enterohepatic circulation. 115 TPE as a treatment for PCT is reported by many authors, but no controlled studies are available.
Other dermatological diseases, such as necrotic xanthogranuloma and scleromyxedema, are not mentioned due to the oncological treatment or the lack of clinical data.
All mentioned TA methods are still technically complicated and very expensive. The costs of the mentioned TA methods vary widely in Germany; for example, the costs for TEP are between 830 and 1,620€, for IA between 2,040 and 2,240€, and for ECP between 1,600 and 2,700€ per treatment. 116 It is the responsibility of the manufacturers to develop simpler and less costly techniques.
Physicians are committed to helping all patients entrusted to them to the best of their knowledge, and this means that medical treatment – and particularly the apheresis processes –must become affordable. This demand represents a great challenge to physicians, politicians, health organizations, and above all to the manufacturers. Industry constantly justifies the high costs with the extensive research and development required. All those involved in the health-care system must intensify their cooperation in this respect.
Summary
TA has been successfully used in varies antibody-mediated diseases. PV is a classic example of antibody-induced immune dermatosis. TPE or IA and ECP are indicated in patients with severe symptoms who either received high doses of conventional agents and/or had an aggressive and rapidly progressive disease. BP is another rare form of subepidermal blistering pemphigus. BP is not as dramatic as other autoimmune diseases with good response of conventional therapy. TPE and IA in combination with immunosuppression are indicated in BP and d-penicillamine–induced pemphigus only in severe cases. Chemotherapy and stem cell transplantation are indicated for more aggressive forms of CTCL. Following the recommendations of the AAC of the ASFA, CTCL has category I and grade 1B for ECP. The advantage of ECP is the relative lack of immune suspension and reduced risk of infection.
In severe cases of progressive scleroderma, dermatomyositis, TEN, psoriasis vulgaris, and HST, TPE or IA can be successful. In these diseases, and in those patients who do not respond to this therapy, the first-line therapy is immunosuppression. Other immunosuppressants, biologics, or TA could act as second-line therapy. TA is only indicated in severe cases of Behcet disease, PG, dermatitis herpetiformis, and herpes gestations as second-line therapy. In the guidelines of AAC of the ASFA, the last four mentioned diseases are not discussed. But for all mentioned diseases, the quotient relevant for cost-effectivity assessment [cost of treatment – cost saved]: [improvement in life quality] must be discussed and calculated exactly by all the persons involved.
Author Contributions
Conceived the concepts: RB. Analyzed the data: RB, DB, RS. Wrote the first draft of the manuscript: RB. Contributed to the writing of the manuscript: RB, RS. Agree with the manuscript results and conclusion: RS, DB. Jointly developed the structure and arguments for the paper: RB, RS. Made critical revisions and approved final version: RB, RS, DB. All authors reviewed and approved of the final manuscript.