
Abstract
Introduction
Cyclopia (alobar holoprosencephaly) (OMIM% 236100) is a rare and lethal complex human malformation, resulting from incomplete cleavage of prosencephalon into right and left hemispheres occurring between the 18th and the 28th day of gestation. Holoprosencephaly occurs in 1/16,000 live births, and 1/250 during embryogenesis. Approximately 1.05 in 100,000 births are identified as infants with cyclopia, including stillbirths. Cyclopia typically presents with a median single eye or a partially divided eye in a single orbit, absent nose, and a proboscis above the eye. Extracranial malformations described in stillbirths with cyclopia include polydactyl, renal dysplasia, and an omphalocele. The etiology of this rare syndrome, which is incompatible with life, is still largely unknown. Most cases are sporadic. Heterogeneous risk factors have been implicated as possible causes.
Case Presentation
A live full-term baby with birth weight of 2900 g, product of cesarean section because of severe fetal bradycardia, was born at Prince Hashem Military Hospital – Zarqa city/Jordan. This newborn was the first baby to a non-consanguineous family, and a healthy 18-year-old mother, with no history of drug ingestion or febrile illnesses during pregnancy. Antenatal history revealed severe hydrocephalus diagnosed early by intrauterine ultrasound but the pregnancy was not terminated because of the lack of medical legitimization in the country. On examination, the newborn was found to have a dysmorphic face, with a median single eye, absence of nose, micrognathia, and a proboscis above the eye, all of which made cyclopia the possible initial diagnosis. Multiple unusual abdominal defects were present that include a huge omphalocele containing whole liver and spleen, urinary bladder extrophy, and undefined abnormal external genitalia, which called for urgent confirmation. Brain MRI was done and revealed findings consistent with alobar holoprosencephaly (cyclopia).
Conclusion
Presentation of cyclopia is not fully exposed and new cyclopian syndromes still can appear. The prenatal diagnosis of cyclopia can be made early by ultrasound, and the awareness of the spectrum of sonographic findings of cyclopia can improve the accuracy of prenatal diagnosis. The legitimization of pregnancy termination for indexed cases in many countries around the world should be revised.
Introduction
Holoprosencephaly refers to a group of disorders arising from failure of normal forebrain development during embryonic life. 1
Three levels of increasing severity are described: 2 alobar holoprosencephaly (cyclopia being the most severe form), with a single brain ventricle and no interhemispheric fissure; semilobar holoprosencephaly with a partial separation; and lobar holoprosencephaly, where the right and left ventricles are separated, but with some continuity across the frontal cortex.
Cyclopia (alobar holoprosencephaly) (OMIM% 236100) is a rare and lethal complex human malformation, resulting from incomplete cleavage of prosencephalon into right and left hemispheres occurring between the 18th and the 28th day of gestation. 3
Holoprosencephaly occurs in 1/16,000 live births,1,4 and 1/250 during embryogenesis. 5 Approximately 1.05 in 100,000 births are identified as infants with cyclopia, including stillbirths. 6
Cyclopia refers to a single midline orbit that contains ocular structures that could be monophthalmic, synophthalmic or anophthalmic. 7
Cyclopia typically presents with a median single eye or a partially divided eye in a single orbit, absent nose, and a proboscis above the eye. Extracranial malformations described in stillbirths with cyclopia include polydactyl, renal dysplasia, and an omphalocele.
The etiology of this rare syndrome, which is incompatible with life, is still largely unknown. Most cases are sporadic. 8 Heterogeneous risk factors have been implicated. Possible risk factors include: maternal diabetes 9 (the only formally recognized environmental factor with a 1% risk and a 200-fold increase in fetal holoprosencephaly), infections during pregnancy (TORCHs),10–14 drugs during pregnancy15,16 (alcohol, aspirin, lithium, anticonvulsants, hormones, retinoic acid, anticancer agents, and fertility drugs), physical agents 17 like ultraviolet light, and chromosomal 3 (mostly trisomy 13) and genetic causes 18 (familial occurrences in twins and in consanguineous marriages have been documented).
Case Presentation
At Prince Hashem Military Hospital – Zarqa city/Jordan, a live full-term baby with birth weight of 2900 g, product of cesarean section because of severe fetal bradycardia, was born. The 18-year-old healthy mother had no history of drug ingestion, or febrile illnesses during pregnancy.
Antenatally the newborn, which was the first baby to a non-consanguineous family, was diagnosed by ultrasound with severe hydrocephalus during early intrauterine life (Fig. 1), but not terminated because of the lack of medical legitimization in the country.

Early intrauterine sonographic findings (single holoventricle with surrounding cerebral tissue and fused thalami).
On examination, the newborn was found to have a pink face and a trunk with peripheral cyanosis. Heart rate was 152 beats/minute and respiratory rate 42/minute, but Apgar score was not calculated because of severe abnormalities. Head circumference was 38 cm, with a dysmorphic face, a median single eye, absence of nose, micrognathia, a proboscis above the eye (Fig. 2), and multiple unusual abdominal defects that include a huge omphalocele containing whole liver and spleen, urinary bladder extrophy, and undefined abnormal external genitalia (Figs. 3 and 4). The newborn expired after 5 hours.
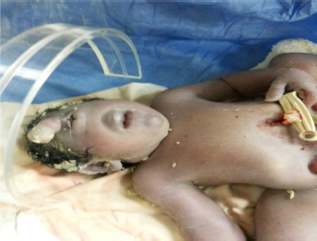
Facial profile of the newborn baby with cyclopia.

The extrafacial associated abnormalities of the newborn baby with cyclopia.
Brain MRI was done, which revealed undifferentiated holosphere of the cerebral parenchyma with a central mono-ventricle, fused thalami, and absence of midline structures, such as corpus callosum and the midline falx cerebri.
Chromosomal analysis and postmortem autopsy were not carried out as consent to these two procedures was not given by the father.
Discussion
During the 4th week of gestation, the neural tube forms the three primary brain vesicles (prosencephalon, mesencephalon, and rhombencephalon) and by the 5th gestational week, the prosencephalon further divides into the telencephalon and diencephalon. The two cerebral hemispheres and the lateral ventricles arise from the telencephalon, whereas the thalami, hypothalamus, and the basal ganglia arise from the diencephalon.
Holoprosencephaly refers to a group of disorders arising from failure of normal forebrain development during embryonic life. There are three forms of holoprosencephaly: alobar, semilobar, and lobar varieties, with alobar holoprosencephaly (cyclopia) being the most severe form and characterized by undifferentiated holosphere of the cerebral parenchyma with a central monoventricle, fused thalami, and absence of midline structures, such as corpus callosum and the midline falx cerebri.19–22 The brain MRI findings in our case are identical to these anatomical findings (Fig. 5).

The MRI findings of the newborn baby with cyclopia (red arrow – single ventricle [the dorsal cyst], blue arrow – fused thalami and absence of midline structures).
Sonography is the most helpful in the prenatal diagnosis of cyclopia.20,23–25 Holoprosencephaly can be expected to present in 16% or more of all cases of fetal hydrocephalus. 26 Even about 17% of fetuses with alobar holoprosencephaly reported by DeMyer 26 and 29% reported by Nyberg23,27 had a nondiagnostic face at delivery, but when holoprosencephaly is suspected by sonography to be the case, careful intrauterine scanning of the face will allow a more definitive diagnosis of cyclopia. One has to remember the well-known phrase, “the face predicts the brain.” Cardinal facial features of cyclopia may include a median single eye or a partially divided eye in a single orbit, absent nose, and a proboscis above the eye. Other facial features are absent philtrum, otocephaly, and astomia or microstomia.
In our case, a severe hydrocephalus was diagnosed early in the first trimester (Fig. 1) but no attention was paid to the facial features, and no fetal MRI to confirm the diagnosis was done. At birth, our case was found to have the typical facial features of cyclopia including a median single eye, absence of nose, micrognathia, and a proboscis above the eye (Fig. 2).
Apart from the facial features of the infant with cyclopia, extrafacial features were reported and could include polydactyly, renal dysplasia, and an omphalocele, all of which can be detected by sonography if looked for them carefully. The presence of extrafacial abnormalities carries a very poor prognosis and almost always associated with stillbirth.23,28
In our case, intrauterine extrafacial features passed undiagnosed.
Most live infants with cyclopia at birth were reported to have the typical facial features but no extracranial ones. During literature review, we found only two reports of live newborn infants with cyclopia having extrafacial malformations in addition to facial features: a live newborn with cyclopia and bladder extrophy was reported by McGahan et al. 23 , and another baby with polydactyly was reported by Corsello et al. 29
The originality of our case is that it is the first case report of a live full-term newborn infant with cyclopia, with typical facial manifestations and three extrafacial malformations, namely a huge omphalocele, bladder extrophy, and ambiguous genitalia (Figs. 2–4).
Moreover, our case is the first reported case of cyclopia with a huge omphalocele containing whole liver and spleen (Figs. 3 and Fig. 4).
And such an amazing combination of cyclopia with ambiguous genitalia as in our case is still not reported anywhere; the finding can open the way for researchers to investigate the relation between congenital adrenal hyperplasia and holoprosencephaly.
Even it is allowed by medical law in many countries to terminate the pregnancy if major congenital abnormalities are detected during pregnancy, but in many other countries it is still not allowed for cultural, religious, and other reasons. In our case, the parents were informed early that their baby has a severe hydrocephalus, but they had no choice to terminate the pregnancy and they were left to suffer the great psychological pain of bearing a deformed fetus till term and delivering alive an abnormal baby who expired a few hours after birth.
This case calls for urgent worldwide legitimization of pregnancy termination in indexed cases.
The last but not the least important fact is that even Holoprosencephally (HPE) is a syndromic malformation with many genetic causes, both with and without an associated chromosomopathy, and chromosomal analysis and postmortem autopsy can add to the diagnosis of cyclopia, but in our case they were not carried out as consent to these two procedures was not given by the father.
Conclusion
Presentation of cyclopia is not fully exposed, and new cyclopian syndromes still can appear. The prenatal diagnosis of cyclopia can be made early by ultrasound and the awareness of the spectrum of sonographic findings of cyclopia can improve the accuracy of prenatal diagnosis. The legitimization of pregnancy termination for indexed cases in many countries around the world should be revised.
Consent
Written informed consent was obtained from our patient's father for publication of this case report and any accompanying images as well as Jordanian Royal Medical Services ethical committee approval.
Author Contributions
GS is responsible for the concept of the case report. GS wrote the manuscript. MK, M. Al-raqad reviewed and edited the manuscript. IMHA, AS, RH reviewed references. All authors read and approved the final manuscript.