
Abstract
Ribosomopathies are diseases caused by alterations in the structure or function of ribosomal components. Progress in our understanding of the role of the ribosome in translational and transcriptional regulation has clarified the mechanisms of the ribosomopathies and the relationship between ribosomal dysfunction and other diseases, especially cancer. This review aims to discuss these topics with updated information.
Keywords
Introduction
As the apparatus of protein synthesis, the ribosome is one of the most precisely constructed and regulated molecular machines in the cell. Alterations in ribosomal components, or in the numerous cellular products with effects on ribosomal structure and function, can cause a heterogeneous class of diseases known as ribosomopathies. Although they all involve ribosomal dysfunction, these diseases differ significantly in mechanism, clinical presentation, and potential for treatment. This diversity corresponds to a developing understanding of the multiple specialized roles of the ribosome in normal function. Recent studies on ribosomopathies have yielded more insight into other diseases, including multiple cancers, and key cellular pathways, notably of the tumor suppressor p53. We will review these developments here, with an emphasis on the molecular mechanisms of disease, and discuss their implications for treatment and further research.
Ribosomes and Ribosomal Proteins: Structure and Function
The ribosome principally consists of ribosomal RNA (rRNA), ribosomal proteins (RPs), and small nucleolar RNAs (snoRNAs). rRNA catalyzes peptide bond formation during protein synthesis; RPs optimize rRNA processing and stabilize the ribosome's final structure; and snoRNAs primarily regulate chemical modifications of other RNAs.1,2 rRNA transcription and assembly with RPs occur in the nucleolus, after which the ribosomal subunit is exported from the nucleus to the cytoplasm, the site of translation. In eukaryotes, regulation of translation occurs primarily at the initiation step and is mediated by eukaryotic initiation factors (eIFs). 3 Most mRNAs are primed for translation via a mechanism by which eIFs recognize either the 7-methylguanylate cap at the 5’ end or the poly-A tail at the 3’ end of the mRNA, facilitating its binding to the translation preinitiation complex (PIC). However, some mRNAs undergo a cap-independent mechanism of initiation, in which the PIC is directly recruited by a nucleotide sequence called the internal ribosomal entry site (IRES) within the mRNA. 4 IRESs were first discovered in viral mRNAs, which are often uncapped; IRESs facilitate translation of viral mRNAs even when eIFs are downregulated, as they commonly are in infected cells.5,6 Eukaryotic genes whose transcripts feature an IRES include those involved in the cellular stress response, and accordingly IRES-dependent translation can promote either apoptosis or cell survival. 7
The ribosome itself has long been thought to play a primarily constitutive role in translation, rather than a regulatory one. However, studies of Escherichia coli in the 1960s showed that alterations in ribosomal structure, such as those induced by the antibiotic streptomycin, can affect translational fidelity.8,9 These findings have been corroborated by mutational analyses of E. coli ribosomes, 10 as well as by structural studies.2,11 Furthermore, analysis of polypeptide chain elongation kinetics has shown that inhibiting the steps that precede mRNA binding preferentially blocks the translation of lower-quality mRNAs, which have smaller initiation rate constants. This represents another mechanism by which translational fidelity can be ensured. 12
More recent investigations have shown that ribosomes and RPs selectively regulate the expression of specific mRNAs, an idea known as the ribosome filter hypothesis. 13 In 2011, Kondrashov et al showed that skeletal patterning defects in tail-short (Ts/+) mice were caused by loss of function in the Rpl38, which resulted in decreased translation in a subset of homeobox proteins. 14 In the same study, quantitative gene-expression profiling of 72 RPs showed heterogeneity of expression levels among different tissues during organogenesis, suggesting that RPs other than Rpl38 can regulate translation in a tissue-specific manner.
Other RPs regulate translation via extraribosomal mechanisms. 15 One such RP, Rpl26, induces translation of p53 by binding to the 5’ and 3’ untranslated regions of p53 mRNA.16,17 It remains uncertain whether ribosomal or extraribosomal effects account for most translational regulations by RPs.
Given the ubiquity of ribosomes and RPs, a paradoxical feature of ribosomopathies is the variety of their phenotypic effects. This variety is explained in part by the emerging concept of “specialized ribosomes,” in which tissue-specific variations in ribosomal structure or function confer regulatory specificity in translation. 18 These variations include the expression of RP paralogs in different tissues,19,20 heterogeneity of RP expression levels during embryogenesis14,21,22 and in adult life, 23 differences in post-translational modifications,24–26 changes in expression of ribosome-associated factors other than RPs,27–30 and heterogeneity in rRNA structure.31–33 The composition of ribosomes and other elements of the translational apparatus can also vary within cells, especially in neurons, where certain RPs and RNAs are selectively enriched in axons or dendrites relative to the soma.34,35 These variations in turn affect the translation of specific subsets of mRNAs. For example, modifications to rRNAs36–39 and to RPs40–42 can selectively impair IRES-dependent translation, and certain RPs appear to be necessary for the translation of some mRNAs but not others. 43 These variations are clinically significant in several ribosomopathies.
Translational and Transcriptional Control: Ribosomes and p53
The transcription factor p53, a key tumor suppressor and regulator of cell fate, is particularly involved in the control of ribosomal function.44,45 p53 can activate or repress transcriptional targets to induce cell-cycle arrest or apoptosis,46,47 and indeed, both cell cycle arrest and apoptosis have been observed in impairments of ribosomal biogenesis. Studies of Bop1, a mouse protein involved in rRNA processing and production of the 60S ribosomal subunit, provided an early indication of the role of p53 in ribosomal dysfunction. A dominant-negative mutation of Bop1 led to p53-dependent cell-cycle arrest that could be relieved by inactivation of p53.48,49 Animal models have since provided further evidence for the connection between ribosomal insufficiency and p53 activation. For example, mice with T-cell-specific heterozygous deletion of the RP gene Rps6 exhibited decreased T-cell-mediated proliferation via a likely p53-dependent response. 50 Mouse embryos with heterozygous Rps6 deletions died at gastrulation, when a p53-dependent checkpoint induced widespread apoptosis. Inactivation of p53 prolonged life briefly, until embryos appeared to die from placental abnormalities and impaired erythropoiesis.50,51 Mutations of other RPs in mice can cause less dramatic phenotypic abnormalities that are also specifically mediated by p53,52 and RP deficiencies in other animals can cause similar p53-dependent effects.53,54
The mechanism by which impaired ribosomal biogenesis activates p53 has become increasingly clear in recent years. 55 Individual RPs, including Rpl11,56,57 Rpl5, 58 and Rpl23,59,60 can migrate from the nucleolus to the nucleoplasm; there, they bind to the protein MDM2, inhibiting its E3 ubiquitin ligase activity and preserving p53 from proteosomal degradation (Fig. 1). The precise effects, however, vary according to the perturbation, and we discuss them in the context of specific ribosomopathies below.

The role of p53 activation in the pathogenesis of certain ribosomopathies. Bars indicate inhibition.
Ribosomopathies: Old and New Mechanisms
Clinical features of the ribosomopathies can include bone marrow failure, developmental abnormalities, and increased risk of cancer. However, ribosomal dysfunction can cause a wide range of signs and symptoms, and presentation and severity can differ dramatically even among patients with the same diagnosis (Table 1).
Clinical characteristics of ribosomopathies.
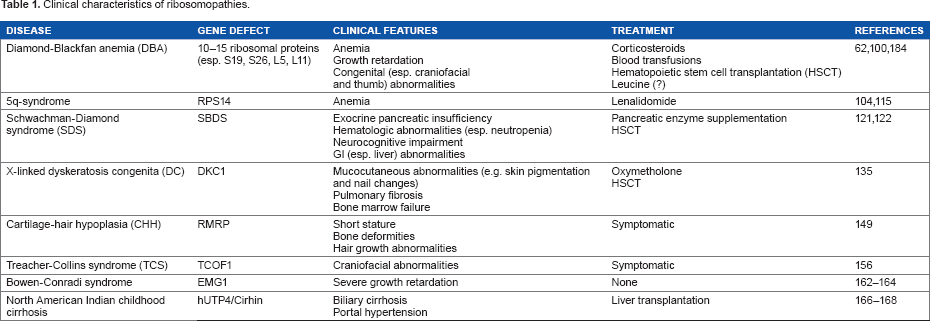
Diamond-Blackfan anemia (DBA)
This disease, a “founding member” of the class of ribosomopathies, 61 presents with pure red cell aplasia in the first year of life as the cardinal symptom. In addition, approximately 40% of patients present with growth retardation or congenital abnormalities of the head, upper limb, kidney, or heart. 62 In 1999, RPS19 was shown to be mutated in DBA, and RPS19 mutations have since been estimated to occur in approximately 25% of cases. 63 Since then, the list of RPs implicated in DBA has expanded to include 10-15 additional candidates, although definitive roles in pathogenesis have not been established for all of them.62,64 Mutations in genes encoding RPs have been identified in 50-70% of patients with DBA. 65 Not all cases of DBA can be attributed to RP dysfunction, however, and a recent study has identified a mutation in the hematopoietic transcription factor GATA1 (which has no known mechanistic links to ribosomes) in two DBA pedigrees. 66 These uncertainties in classification may resolve with improved understanding of the molecular basis of the disease.
All RP mutations observed in DBA are heterozygous, supporting the idea that homozygous RP mutations are lethal to embryos. This hypothesis has been confirmed in some animal models of DBA.52,67 Mutations in the RPS19 gene have been identified as a possible cause of hydrops fetalis, suggesting that more severe RP mutations than those observed in DBA patients may not come to clinical attention. 68 More than 200 mutations in DBA-associated genes have been cataloged in DBA patients, 69 and as a result, the phenotype of DBA patients is also highly variable. Most cases of DBA are sporadic in transmission, but incomplete penetrance and variable expressivity are observed even in familial cases associated with the same mutation. 70 Array comparative genomic hybridization studies have also identified genomic deletions of RP loci in mutation-negative patients. 71 Some genotype-phenotype correlations have been established by studies of patient cohorts; for example, mutations in RPL5 and RPL11 are associated with oral cleft and thumb abnormalities, respectively,72,73 while RPL35A mutations are associated with genitourinary malformations. 69
Given the diverse etiologies and presentations of DBA, understanding of its pathogenesis is incomplete. This state of affairs is further complicated by the fact that animal models to date have not fully replicated the DBA phenotype.74–76 RP deficiencies are associated with global decreases in translation in cells of both hematopoietic and non-hematopoietic lineage, an observation that may account for general features of DBA such as small stature.77–79 RP “crosstalk” has been observed in some studies, where removal or knockdown of an RP promoted the recruitment of other RP transcripts to polyribosomes for translation, but this enhanced recruitment could not compensate for decreased translational efficacy.42,80
The role of p53 in the pathogenesis of DBA is also incompletely understood. In vitro RNAi knockdown of RPS19 in human CD34+ hematopoietic progenitor cells replicates the defective erythropoiesis of DBA81,82 and is associated with p53 accumulation in the erythroid lineage; in this setting, restoration of RPS19 expression and p53 inactivation both rescue the DBA phenotype. 83 RPS19 knockdown interferes with ribosomal biogenesis in yeast 84 and in mammalian cells, leading to p53 activation.85,86 More generally, knockdown of some RPs can relieve miRNA-mediated repression of translation, again via p53 activation. 87 RPL5 and RPL11, the two RPs that are responsible for a plurality of observed non-erythroid phenotypes when mutated in DBA, have the specific function of transporting 5S rRNA to the developing 60S ribosomal subunit and forming a complex that inhibits Mdm2.88,89
It has been postulated that hypersensitivity of erythroblasts to p53 or high requirements for protein synthesis during erythropoiesis account for the central role of p53 in the pathogenesis of DBA78,83 (Fig. 1). Intriguingly, a recent study of CD34+ cells from DBA patients suggested that the effect of p53 activation can vary depending on the affected RP; RPS19 mutations decreased proliferation and cell-cycle arrest but had little effect on differentiation and apoptosis, while RPL11 mutations delayed differentiation and increased apoptosis. 90 There is also evidence for p53-independent mechanisms in the pathogenesis of DBA and especially of the erythroid defect. In an RPS19-deficient zebrafish model of DBA, for example, inhibition of p53 rescued the morphological abnormalities, but not the erythropoietic defect. 91 RP knockdown in a p53-deficient mouse erythroblast line increased transcription but decreased translation of Bag1 and ∗∗∗Csde1, two proteins that are essential for erythroid differentiation. 42 Translation of Bag1 and Csde1 is cap-dependent and IRES-mediated, and impaired IRES-mediated translation is also observed in models of other ribosomopathies.36,39 RPS19 is also thought to play a role in the regulation of alternative splicing; 92 enhanced alternative splicing and decreased translation of FLVCR1, an erythroblastic heme exporter that plays a role in erythroid differentiation, have been observed in hematopoietic stem cells of RPS19-mutated DBA patients. 93
Corticosteroids, which were first shown to be efficacious in DBA treatment in 1951, remain the first-line therapy today. Activation of the glucocorticoid receptor promotes erythroid proliferation,94,95 and it is believed that corticosteroids exert a general antiapoptotic effect among erythroid precursors and downregulate the expression of non-erythroid genes.82,95 Corticosteroids also increase sensitivity to erythropoietin, levels of which are often elevated in DBA patients. 96 Leucine, which stimulates translation via the mTOR pathway, has been shown to improve hematopoiesis in some animal models of DBA,97,98 as well as in a small clinical trial; 99 larger trials are underway. Hematopoietic stem cell transplantation, the only curative treatment for the anemia, is currently considered for certain patients and is likely to be further extended as outcomes improve.100,101
5q-syndrome
This disease, first reported as a refractory anemia with a distinct karyotype in 1974, shares clinical and pathologic characteristics with DBA. Now considered a subtype of myelodysplastic syndrome (MDS), it is defined cytogenetically, as a de novo deletion of the region between bands q21 and q32 of chromosome 5. 102 Although 5q deletions are also observed in other cases of MDS and acute myeloid leukemia (AML), the region involved in 5q-syndrome is known to be distinct. 103 Clinically, 5q-syndrome presents predominantly among females, with a more indolent course, lower risk of progression to AML, and more favorable prognosis than other subtypes of MDS. Blood findings include macrocytic anemia with hypolobulated megakaryocytes and normal or elevated quantities of platelets, with <5% blasts in both bone marrow and peripheral blood. 104
Haploinsufficiency of RPS14 is critical to the pathogenesis of 5q-syndrome. shRNA knockdown of RPS14 in human CD34+ hematopoietic stem cells has been shown to replicate the erythroid defect, which could in turn be rescued by RPS14 overexpression. 105 This finding has been confirmed in a mouse model of the disease, which further suggested a p53-dependent mechanism for the anemia, 106 and recent studies in cancer cell lines have confirmed that RPS14 exerts both p53-dependent and p53-independent tumor suppressor effects107,108 (Fig. 1). Other genes in the deleted region also appear to play a role in the pathogenesis of 5q-syndrome. These include the tumor suppressor gene SPARC, which has antiproliferative and anti-angiogenic effects; 109 miR-145 and miR-146a, whose deletion may contribute to the thrombocytosis; 110 and the candidate tumor suppressors EGR1, CTNNA1, and CDC25C, among other genes.111–114
5q-syndrome is characterized by a striking therapeutic response to lenalidomide, a thalidomide analog with fewer side effects and increased selectivity115,116 (Fig. 1). Lenalidomide is also used as a treatment for multiple myeloma; in this setting, it exerts immunomodulatory and antiangiogenic effects, in addition to directly inhibiting the proliferation of neoplastic cells. 117 Although its mechanism of action in 5q-syndrome is incompletely understood, it has been shown to promote erythroid differentiation, 118 upregulate SPARC, 109 inhibit the cell-cycle regulators Cdc25C and PP2Acα, 114 induce cell death by blocking cytokinesis, 119 and promote degradation of p53. 120
Shwachman-Diamond Syndrome (SDS)
This rare disorder is clinically characterized by exocrine pancreatic insufficiency and hematologic abnormalities, most commonly neutropenia.121,122 Skeletal and neurocognitive abnormalities have also been observed, and there is increased risk of neoplastic transformation, particularly to MDS and AML. Although the diagnosis is usually made in the first few years of life, SDS can also present among older children and even adults. The presentation can vary widely, although confirmation of pancreatic and hematologic dysfunction is necessary to establish the clinical diagnosis. 121
Approximately, 90% of patients with a clinical diagnosis of SDS carry biallelic mutations in the SBDS gene. The SBDS protein is highly conserved among archaea and eukaryotes; in mammals, it has been ascribed functions in key cellular pathways, including mitotic spindle stabilization,123,124 DNA metabolism and stress responses, 125 reactive oxygen species regulation, 126 and actin-dependent processes such as chemotaxis,127,128 in addition to ribosomal biogenesis.129,130 Mouse models have indicated roles for SBDS in both hematopoietic and stromal cells of the bone marrow.131,132 Because of this multiplicity of functions, the role of SBDS in the pathogenesis of SDS is unclear, and the classification of the disease as a ribosomopathy has been somewhat controversial. However, recent studies have established that the role of SBDS in ribosomal biogenesis is conserved in Saccharomyces cerevisiae and other eukaryotes as well as mammals. In particular, two recent studies showed that SBDS interacts with the GTPase EFL1 to catalyze the removal of eIF6 from the 60S ribosomal subunit, a key step in the formation of actively translating ribosomes.133,134 The latter of these studies also demonstrated defects in ribosomal subunit joining in lymphoblasts from SDS patients, supporting the idea that ribosomal dysfunction plays a role in the disease. 134
Dyskeratosis Congenita (DC)
This genetically and clinically heterogeneous disease is classically associated with mucocutaneous abnormalities, pulmonary fibrosis, bone marrow failure, and predisposition to cancer. 135 DC can be caused by mutations in any of approximately 10 genes, each of which is associated with telomerase function or telomere integrity;136,137 in addition, Hoyeraal-Hreidarsson syndrome, a severe variant of DC whose symptoms include cerebellar hypoplasia, immunodeficiency, and enteropathy, has recently been shown to be caused by mutations in the regulator of telomere elongation helicase Rtel1.138,139 Because of the clear role of telomere defects in the pathogenesis of DC, authors have tended to classify it among other diseases that cause telomere shortening, including ataxia-telangiectasia, Bloom syndrome, and Fanconi anemia. 136 Indeed, the clinical phenotype of DC is consistent with premature aging and loss of cells in high-turnover tissues. 140
Defects in rRNA processing have been suggested to play a role in the X-linked form of DC, which is caused by mutations in the DKC1 gene. DKC1 encodes the dyskerin protein, which is found not only in the telomerase complex but also in H/ACA ribonucleoprotein complexes, where it is involved in rRNA pseudouridylation.141,142 The functional consequences of this ribosomal defect remain controversial, but there is evidence that it leads to decreased translational fidelity39,143 and impaired control of IRES-mediated translation.36,144,145 This dysregulation of translation may contribute to increased susceptibility to cancer in DC patients.146–148
Cartilage-Hair Hypoplasia (CHH)
This disorder, most commonly observed in Old Order Amish and Finnish populations, is clinically characterized by short stature, hair growth abnormalities, and bone deformities that can be detected radio-graphically. 149 Anemia and immunodeficiency can also occur, among other symptoms. The disease is autosomal recessive in transmission, but the observed phenotypes are widely variable even within families. Some authors have described a spectrum of disorders including metaphyseal dysplasia without hyper-trichosis, CHH, and anauxetic dysplasia (AD), ranging from least to most severe. 149 There is increased risk of cancer, particularly non-Hodgkin lymphoma and basal cell carcinoma. 150
CHH and other disorders on the CHH-AD spectrum are caused by mutations in the untranslated RMRP gene, which encodes the RNA component of the RNase mitochondrial RNA-processing (MRP) complex. 151 RNase MRP RNA, which is classified as a snoRNA, plays several roles in the normal cell, and as with dyskeratosis congenita, the role of ribosomal dysfunction in the pathogenesis of CHH is uncertain. Nme1, the yeast homolog of RMRP, is involved in rRNA processing.152,153 On the other hand, in mammalian cells, RNase MRP RNA forms complexes with the catalytic subunit of telomerase reverse transcriptase–-one of the genes mutated in some cases of DC–-to produce double-stranded RNA that can be processed into small interfering RNAs (siRNAs). 154 Targets of these siRNAs include genes involved in skeletal development, hair development, and hematopoietic differentiation, among others, suggesting that disturbed siRNA production may be the primary pathogenic mechanism in CHH. 155
Treacher-Collins Syndrome (TCS)
This disease, which is autosomal dominant in transmission, is characterized by craniofacial defects involving structures derived from the first and second branchial arches. 156 Most cases of TCS are caused by mutations in the TCOF1 gene, whose protein product, named treacle, is involved in rRNA transcription and processing and is most strongly expressed in neural crest cells of the branchial arches during the embryonic period.157–159 As in other ribosomopathies, neural crest cell hypoplasia in TCS has been attributed to apoptosis caused by impaired ribosomal biogenesis. 158 In a TCOF1-haploinsufficient mouse model, inactivation of p53 was sufficient to prevent craniofacial abnormalities 160 (Fig. 1). Mutations in subunits of RNA polymerases I and III, which transcribe distinct rRNA sub-units, have also been identified among TCS patients without TCOF1 mutations. 161 Unlike other ribosomopathies, TCS is not associated with hematologic abnormalities, in agreement with the idea that ribosomes carry out specialized activities in different tissues but leaving open the question of how these activities are regulated.
Other Ribosomopathies
Ribosomal dysfunction has been implicated in the pathogenesis of several other diseases. Bowen-Conradi syndrome, an extremely rare disorder that causes severe growth retardation and death in early childhood, is caused by mutations in EMG1, which encodes a pseudouridine methyltransferase involved in ribosomal biogenesis.162–165 North American Indian childhood cirrhosis, which affects children in a small population in Northwestern Quebec, is caused by mutations in hUTP4/Cirhin, which is involved in the synthesis of 18S rRNA.166–168 Haploinsufficiency of ribosomal protein SA, a component of the 40S subunit, was recently demonstrated in a majority of studied cases of isolated congenital asplenia. 169 Impaired ribosomal biogenesis has been proposed to play a role in certain cases of common variable immunodeficiency, possibly as an atypical presentation of more canonical ribosomopathies such as DBA and SDS.170,171 Mutations in eIFs that may disrupt translational machinery and stress responses are observed in vanishing white matter disease172–174 and some cases of autism spectrum disorders.175–177
Ribosomal Dysfunction and Cancer
As our survey indicates, susceptibility to cancer is a common symptom of ribosomopathies. Indeed, at the cellular level, dysregulation of translation has been proposed as a common pathway for cancer progression. 178 The mechanistic link may appear paradoxical, since cellular proliferation in tumors is generally associated with an increase in ribosomal biogenesis and translation, and several cancers induce overexpression of RPs. 179 In the case of X-linked DC, the selective impairment of IRES-mediated translation results in decreased expression of tumor suppressors, such as p53, promoting proliferation and tumor growth.36,37,144 However, evidence for increased IRES-mediated translation of other proteins in X-linked DC suggests that this description may be too straightforward. 145 Putative mechanisms of cancer progression in other ribosomopathies are not as well studied, but may involve regulatory crosstalk between RPs and other components of the translational apparatus with known oncogenic pathways, including those involving c-Myc180–182 and mTOR. 183 The widespread activation of apoptosis observed in several ribosomopathies raises the possibility of selective pressure in favor of cellular clones in which p53 or other regulatory effectors are mutated or otherwise dysregulated. These explanations are preliminary, however, and the role of ribosomes in disease progression remains a largely open problem in cancer research.
Conclusion
With the notable exception of 5q-syndrome, treatment for most ribosomopathies has been symptomatic, with hematopoietic stem cell transplantation ultimately indicated in cases of progression to cancer or severe bone marrow failure. The possibility of treatment via novel pathways, such as inactivation of p53, has been proposed by some authors, 45 but the potential of such therapies may be limited by the heterogeneity of disease mechanisms, as well as by the counterbalancing risk of cancer owing to the loss of p53 surveillance (Fig. 1). In spite of these challenges, the emerging diversity of ribosomal functions sheds light on the molecular basis of the ribosomopathies and informs the development of potential treatments. This improved understanding, as well as improved outcomes in established treatment protocols, is a sign of clinical progress against this complex class of diseases.
Author Contributions
Wrote the first draft of the manuscript: HN. Contributed to the writing of the manuscript: HN, JK, XZ, WL, SXZ, HL. Agree with manuscript results and conclusions: HN, JK, XZ, WL, SXZ, HL. Jointly developed the structure and arguments for the paper: HN, JK, HL. Made critical revisions and approved final version: HL. All authors reviewed and approved of the final manuscript.
Footnotes
Acknowledgments
We thank Ramzi Nakhoul for editorial assistance and all the members of the Lu laboratory for active discussion.