Significant research has been devoted to predicting diagnosis, prognosis, and response to treatment using high-throughput assays. Rapid translation into clinical results hinges upon efficient access to up-to-date and high-quality molecular medicine modalities.
We first explain why this goal is inadequately supported by existing databases and portals and then introduce a novel semantic indexing and information retrieval model for clinical bioinformatics. The formalism provides the means for indexing a variety of relevant objects (e.g. papers, algorithms, signatures, datasets) and includes a model of the research processes that creates and validates these objects in order to support their systematic presentation once retrieved.
We test the applicability of the model by constructing proof-of-concept encodings and visual presentations of evidence and modalities in molecular profiling and prognosis of: (a) diffuse large B-cell lymphoma (DLBCL) and (b) breast cancer.
The goal of Molecular Medicine is to diagnose and find treatments for human diseases by the application of tools of molecular and cell biology (Sobie et al. 2003). In recent years, researchers have begun to link tissue molecular profiles—such as gene expression information—of individual patients to relevant disease outcomes such as diagnosis (Quackenbush, 2006), prognosis (Ntzani and Ioannidis, 2003), and response to treatment (Ross and Ginsburg, 2003). Knowledge discovered from large-scale genomic and molecular biology data is already being put to clinical use (van't Veer et al. 2002) and several clinical studies are in the development or validation phase (Simon, 2005).
The field of pharmocogenomics, for example, applies whole genome analysis technologies to predict drug treatment response and adverse drug reaction susceptibility based on individual genetic variability (Marsh and McLeod, 2006; Ross et al. 2004). For instance, an inherited genetic trait places some individuals at risk for adverse drug reactions (diarrhea, neutropenia) to the antineoplastic drug irinotecan (Ando et al. 2000; Ciotti et al. 1998; Innocenti et al. 2004). Individuals with the most common variant allele (UGT1A1*28) have lower expression levels of an enzyme that deactivates irinotecan. The FDA requires that the related genotype and dosing guideline information be included in the irinotecan package insert (Food and Drug Administration 2008). Other mutations are associated with a good clinical prognosis (Bell et al. 2005) and positive response to certain classes of drugs (Lynch et al. 2004). A listing of drug-related genomic biomarkers is available on the FDA website (Food and Drug Administration, 2008).
In a typical scenario, a molecular assay is performed on tissue obtained from a patient. Then, a decision model computes, based on the assay results, the “predicted” clinical outcome of the patient's disease. For example, the U.S. Food and Drug Administration approved in February of 2007 the first high-dimensional molecular test to predict the recurrence of breast cancer within five to ten years. Many similar tests are expected to follow (Couzin, 2007).
Discovering clinically significant knowledge from large-scale genome and molecular biology information is a complicated scientific process that draws from multiple overlapping sources of data describing complex interactions at the genomic, proteomic, or other “omic” levels. High throughput “omic” experimental methods generate data that can have hundreds or even hundreds of thousands of data-points per sample. Such data are difficult to process manually and require sophisticated computation. Decision models that process the resulting data are also complex and draw from a variety of disciplines including biostatistics and machine learning. Furthermore, there is great variability in the methods that evaluate these predictive models' validity, generalizability, and supporting evidence (Simon, 2005).
For advances in molecular medicine to come to clinical fruition, it is crucial for clinical and translational researchers to have access to relevant, up-to-date, and correct information about known molecular medicine modalities (Mathew et al. 2007), such as research datasets, research methods, known and validated decision models, and related evidence. Therefore the important problem of retrieving and organizing the vast amount of information issued from molecular medicine research needs to be addressed. The inherent complexity of this domain and the fast pace of scientific discovery make this problem particularly challenging.
Problem Statement
Our goal is to develop a general purpose information retrieval system that satisfies the following two requirements:
The system should be able to index, retrieve and organize most methods of molecular profiling, most forms of predictive computational models, many types of clinical outcome, as well as supporting evidence and computational resources.
The knowledgebase needs to be comprehensive and up to date. This requires simple, cheap, fast, and scalable methods to build the knowledge base and to keep it current. To keep up with the rapid pace of discovery in clinical bioinformatics, these methods have to be automated or semi-automated in the worst case.
For this system to support the first requirement, its underlying knowledge representation formalism has to convey the semantic complexity of the clinical bioinformatics domain; on the other hand, the underlying formalism has to be simple enough to support the second requirement of relying on scalable automated methods. The problem, therefore, is to develop a framework and semantic model that balance these two requirements.
This system will also have to accommodate a wide range of query types. Consider the following query examples to be posed by clinicians and/or clinical and translational researchers:
Example Query 1: “What models exist that predict the response to the chemotherapy regiment (CHOP) in patients with diffuse large B-cell lymphoma (DLBCL)?” In this query, the following entities are specified: “disease” is specified as “DLBCL”; “clinical outcome” is specified as “response to CHOP”. Notice that this question leaves the specific method of “molecular profiling” open. This query might be posed by an oncologist looking for up-to-date knowledge to guide her choice of treatment strategy for her DLBCL patient.
Example Query 2: “What models exist that predict response to the chemotherapy regiment (CHOP) based on gene expression profile?” This query does not specify the type of cancer, it does, on the other hand, restrict all desired models to those based on gene expression data. This query may be posed by a researcher in pharmacogenomics looking to correlate the expression of specific genes with the biological function of specific drugs.
Example Query 3: “What papers have compared multiple supervised learning methods for the prediction of cancer diagnosis based on gene expression data using a cross validation method?” This query could be posed by a clinical researcher in possession of a gene expression dataset who is looking for proven methods to build and validate models for diagnosing prospective cancer patients using gene expression microarrays. Notice that in this query, the specific disease and the specific outcome are not specified. Only the type of outcome is specified as “diagnosis”. Also notice that this query specifies classes of algorithms (“supervised learning”) and validation methods (“cross-validation”) rather than individual methods.
Example Query 4: “What datasets originating from breast tumor samples contain mass spectrometry data and contain clinical survival data?” This is a specific query by someone who is interested in building and testing models that predict survival in breast cancer based on raw mass spectrometry data.
These queries require the search and retrieval of a multiplicity of molecular medicine modality object types including but not limited to documents, which are the focus of traditional information retrieval problems. Our envisioned system is intended to represent and retrieve four different types of objects relevant to clinical bioinformatics:
Papers: A published paper is the primary unit of scientific communication. Individual papers or groups of papers describe the methods and results of high throughput molecular medicine research.
Datasets: In many cases, researchers publish their data in the public domain (Broad Institute 2005). Often, that data is utilized by other researchers seeking to develop new and improved analysis methods, to test novel hypotheses, or simply to reproduce or validate the published results.
Algorithms/Software: Research laboratories that develop data analysis methods often publish implementation of the algorithms that they have developed and applied (Broad Institute 2008).
Models: Predictive computational models are produced by the application of algorithms on research datasets. Predictive computational models provide a “decision” based on molecular assays and clinical data obtained from a single patient. The predictive computational model's decision (output) may then be used for the clinical management of the respective patient, for example to help determine the choice of effective therapy. Ideally the process of decision model formation includes rigorous statistical validation to ensure that the utility of a given decision model can generalize to a wider population.
Related Work
Existing information retrieval systems specialized for molecular medicine modalities store and organize only related subsets of clinical bioinformatics research information. For example, PharmGKB (Altman et al. 2003; Oliver et al. 2002) is a database that links genomic variability, mostly accounted for by single nucleotide polymorphisms (SNPs), with phenotypes relating to pharmacokinetics, pharmacodynamics, or therapeutic clinical outcomes. Information is organized in PharmGKB by gene, drug, disease, publications, or datasets. ONCOMINE (Rhodes et al. 2004; Rhodes et al. 2007), a database and web-based analysis and visualization tools, is restricted to cancer-related gene expression microarray experimental results. Datasets in Oncomine are profiled (annotated) by cancer and tissue types, by experimental methods, and by the types of gene expression differential analysis performed on these datasets, e.g. comparing gene expression differentials across different prognosis groups or across different histological subtypes. Oncomine provides links to the original datasets as well as analysis tools for (clinical) differential analysis of these datasets, but does not store or classify the applied algorithms or inferred models that were reported in the original publications. The Gene Expression Omnibus (GEO) (Barrett et al. 2007; Edgar, Domrachev, and Lash, 2002), is a resource developed by the NCBI as a MeSH-indexed public repository of microarray and other forms of high-throughput “omics” data submitted by the scientific community. Sources of data in GEO include gene expression microarrays, ArrayCGH, SNP Arrays, Serial Analysis of Gene Expression (SAGE), Massively Parallel Signature Sequencing (MPSS), protein arrays, and mass spectrometry. Information in GEO is organized by series (study-centered data) or by individual genes. Many journals require that gene expression results be submitted in MIAME-compliant format (Brazma et al. 2001) to the GEO prior to publication (Ball et al. 2004). Some of the series in GEO are further curated and stored as datasets with more structured annotations (relevant citations, organisms) and the possibility to perform online data analysis. The Biometric Research Branch at the NCI has developed array analysis tools for gene expression data, and provides a hand-curated archive of human cancer gene expression datasets (Simon and Zhao, 2008). The Rembrandt (National Cancer Institute, 2005) repository is highly annotated for clinically-oriented outcomes but is restricted to brain-cancer-related molecular research data.
In addition to the above, formalisms and tools have been developed to allow genomic and proteomic researchers to ask questions of diverse data repositories. Such cross-database information queries benefit from standard and controlled representation of domain knowledge (Aitken, Webber and Bard, 2004; Smith et al. 2005). By standardizing and controlling domain concepts, ontologies such as the NCI Thesaurus (Sioutos et al. 2007), the Gene Ontology (GO) (Ashburner et al. 2000) and the Clinical Bioinformatics Ontology (REFSEQ) (Hoffman, Arnoldi and Chuang, 2005) support interoperability between clinical bioinformatics repositories. Ontology-based frameworks, such as the RAD/RAPAD Study Annotator (Manduchi et al. 2004), the Functional Genomics Experiment Model (Jones et al. 2004; Jones et al. 2006), and the Ontofusion system for biomedical database integration (onso-Calvo et al. 2007; Perez-Rey et al. 2006), support cross-database queries. Description logic(DL)-based languages (Baader, 2003), such as the Web Ontology Language (OWL) (McGuinness and van Harmelen, 2004) are popular means of formal ontology representation. DLs can be used for conceptual modeling, information integration, and support for semantic query mechanisms. As such, none of these resources provide a general-purpose information retrieval framework for clinical bioinformatics predictive models and related modalities as befits our goal.
Model Formulation and Proof of Concept
Model: Objects, indexing scheme, and queries
We developed an information retrieval model to support our intended system by examining use cases that mimic the queries introduced above in the domains of diffuse large B-cell lymphoma (DLBCL) and breast cancer. The model is described in the context of the task of retrieving research information from the semantically complex clinical bioinformatics domain of gene expression microarrays in the diagnosis and treatment of DLBCL.
Initially, we conducted manual literature reviews for papers that describe this domain. We noted the different objects that were described in the papers that were reviewed, i.e. by identifying Algorithms, Datasets, or Models described in each Paper. Conceptually, the objects in the knowledgebase are all the Papers, and the union of all Algorithms, Datasets, and Models that are described by the Papers. An Algorithm, a Dataset, or a Model can be referenced in more than one Paper.
Further examination of these objects revealed that each can be described by at least one Context that specifies the following elements in a tuple: <Disease, Population, Purpose, and Modality=. For example in the Paper by Wright et al. (Wright et al. 2003), a Model that predicts the molecular subtype of DLBCL was produced and validated by applying the Algorithm “Bayes Classifier” on two gene expression Datasets. The five objects (1 Paper, 1 Algorithm, 2 Datasets, and 1 Model) can each be annotated with the following Context: (Disease = DLBCL, Population = Human Patients, Purpose = Predict Molecular Subtype, Modality = Gene Expression Microarray).
A query to the knowledgebase should then return a subset of the objects in the knowledgebase. A simple enumeration of Papers, Algorithms, Datasets, and Models that relate to gene expression microarrays in the context of DLBCL is shown in the left side of Figure 1. We also realized that a query can be represented as a partial or complete Context. For example, the Contexts represented by the example queries above are shown in Table 1. Queries 1–3 specify partial Contexts, and Query 4 specifies a complete Context. A quick and simple indexing scheme can be achieved by using a set of canonical terms for each of the Context elements, and then indexing each of the objects with at least one complete Context tuple. Objects are retrieved when their Context elements match the Context elements specified in the query.
An overview of how the information retrieval model will be applied to the DLBCL use case. Left side: After specifying the desired query parameters (Context, Quality Filtration), the system will return a potentially large result set of molecular medicine modality objects. This enumerated set of objects is the raw result. Please refer to the subsection “Model: Objects, Indexing Scheme and Queries,” last two paragraphs. Right side: One or more subsets of the raw result may then be selected by the user for visualization and organization based on the relationships between these objects. The subsection “Model: Object Relationships and Quality Filters” elaborates on this process. The full details of the DLBCL use case are mentioned in the subsection “Proof of Concept: Diffuse Large B C-Cell Lymphoma”. Three subsets of objects from the DLBCL domain along with their relationships are organized pictorially according to our model in Figures 2, 3 and 4.
Contexts partially or completely specified by the example queries in the problem statement section above.
Query #
Disease
Population
Purpose
Modality
1
DLBCL
Human Patients
Response to CHOP Regimen
–
2
–
–
Response to CHOP Regimen
Gene Expression
3
–
–
Diagnosis
Gene Expression
4
Breast Cancer
Human Patients
Predict Survival
Mass Spectrometry
We conducted a broad search for DLBCL gene-expression-related objects, by placing a query as in Figure 1 that specified the following Context: (Disease = DLBCL, Modality = Genomic). In the following section we will discuss three clinical bioinformatics scenarios that involve a subset of DLBCL gene-expression-related objects. The scenarios were encountered when we analyzed the set of manually collected objects that satisfied this Context.Figures 2–4 will provide a pictorial representation of these scenarios.
A pictorial representation of the first three widely cited Papers relevant to the DLBCL use case along with the Datasets, Algorithms, and Models that were described in these Papers. Identifying and presenting relationships between these objects is important for the semantic organization of this domain. These relationships are represented by edges connecting the different objects. For example, the three Papers each describe how Algorithms were applied to Datasets to produce decision Models. We identify this class of ternary relationship as Run_on_Produce (Produce in the figure for simplification). Furthermore, the Shipp (Shipp and others, 2002) and the Rosenwald (Rosenwald and others, 2002) Papers describe how the rightmost and leftmost predictive Models (respectively) were validated using the Datasets that they had assayed. This scenario is detailed in the subsection “Proof of Concept: Diffuse Large B-cell Lymphoma,” paragraphs 1–3.
This figure shows the objects and relationships that surround the production and external validation of a Bayes-classifier Model as described in the Wright et al. (Wright and others 2003) Paper and explained in the subsection “Proof of Concept: Diffuse Large B-Cell Lymphoma”, paragraph 4. The Model (bottom center) was produced by applying the Bayes-classifier Algorithm to the lymphochip Dataset (left). The Model was internally validated (left side arc) using that Dataset which was split into independent training and testing sets. It was then externally validated (right side arc) using another independent Dataset that was assayed and described in a previous Paper (right). It is important to represent and identify this type of scenario in which higher quality Models are produced, i.e. Models that generalize across different Datasets and, in this case, across different molecular assay platforms (oligonucleotide vs. cDNA).
This figure describes how an Algorithm (PCA + SIR) was described by the Li et al. (Li, 2006) Paper. This Algorithm was benchmarked using two independent Datasets that were assayed and described by previous Papers, and one Dataset produced by Monte Carlo simulation. The Models that were produced by the application of this Algorithm on these Datasets were validated internally using one independent split of the respective Datasets. This scenario is commonly encountered in methodological research aimed at developing and benchmarking new classification Algorithms. Please refer to subsection “Proof of Concept: Diffuse Large B-Cell Lymphoma,” paragraph 5.
Proof of concept: Diffuse large B-cell lymphoma
DLBCL is the most common form of non-hodgkins lymphoma in adults. Historically, less than half of DLBCL patients are cured by chemotherapy (Vose, 1998). It was suggested early on that DLBCL actually comprises several diseases that differ in responsiveness to chemotherapy. A pioneering paper by Alizadeh et al. in 2000 (Alizadeh et al. 2000) applied bioinformatics methods to investigate this hypothesis. They measured gene expression levels in lymphoid tissue collected from a variety of healthy and sick individuals. The microarray platform used, called “lymphochip,” measured mRNA levels by hybridization on cDNA spots. The cDNA gene library on the lymphochip was deliberately designed to include genes known to be expressed in lymphoid tissue. The resultant Dataset, which consisted of around 17 thousand gene expression analytes for 128 samples, was analyzed using an unsupervised hierarchical clustering Algorithm. Based on the hierarchical clustering results, multiple decision Models were generated that either related to the biological behavior of DLBCL or to the clinical outcome of patients suffering from DLBCL (See Fig. 2). In the former category, the decision Models seemed consistent with the following hypotheses: (1) That DLBCL can be distinguished based on gene expression data from follicular lymphoma (FL), another form of lymphoma; (2) That there are two molecular subtypes of DLBCL; and (3) That one subtype's molecular signature resembles that of activated peripheral B-cells (APB-like) whereas the other's signature resembles that of B-cells found in the germinal centers of lymph nodes (GC-like). The resultant clinical decision Model of this study was that DLBCL samples that clustered in the GC-like category had better survival than those that clustered in the APC-like category.
Two subsequent studies attempted to further investigate and validate the hypotheses that were reported in the Alizadeh Paper. See Figure 2 for a graphical view of the objects and relationships that were reported in these three Papers. Rosenwald et al. used the same microarray platform, the lymphochip, to collect data from 240 patients with DLBCL (Rosenwald et al. 2002). In this study, two Algorithms were used. An unsupervised hierarchical clustering Algorithm was used in a similar way to that described in the Alizadeh paper. However, three resultant hierarchical clusters (molecular subtypes) were found and labeled: “Activated B-Cell-like”, “GC-B-Cell-like”, and “Type 3”. The second Algorithm relied on multivariate regression techniques to construct a clinical survival prediction Model based on (so-called) gene expression scores. The decision Model was derived from a Dataset of 160 patients and was validated on the remaining 80 patients. This decision Model instance was compared to another widely used clinical predictive Model, the “International Prognostic Index” (IPI) (The International Non-Hodgkin's Lymphoma Prognostic Factors Project 1993), that predicts clinical outcome based only on clinical parameters. Molecular and clinical data were reported as independent factors in predicting clinical outcomes.
In a third study, by Shipp et al. (Shipp et al. 2002), gene expression was measured in tumor samples from 58 DLBCL patients receiving the CHOP chemotherapy protocol, and from 19 FLpatients. In this study, however, oligonucleotide-based microarrays were used instead of the cDNA-based lymphochip. Supervised learning methods (Algorithms) were used to construct two predictive classifiers (decision Models): one associated with the biological hypothesis that DLBCL can be distinguished from FLbased on gene expression data, and another associated with the clinical hypothesis that gene expression data can predict the clinical outcome of DLBCL. The latter decision Model was also compared to the IPI clinical predictive Model, and in this study as well, molecular and clinical data were found to be independent factors in predicting outcomes. A more rigorous cross validation method was used to validate the models produced by this study. In this paper, the previous claims about molecular sub-types were put to test. The same unsupervised hierarchical clustering Algorithm was applied on their dataset1 to cluster the samples. Two molecular subtypes did emerge, and they did show “APB-” and “GC-” B-cell-like expression patterns. However, survival was not found to be different between the two groups.
Notice that the oligonucleotide sequences on the microarrays platform of this study were matched through their annotations to the cDNA genes in the “lymphochip” platform used in the other studies. Only the sequences that matched were used in this clustering technique. That's why the ternary relationship apply-on-to-produce has an asterisk in Figure 2.
Wright et al. (Wright and others 2003) wanted to reconcile the results from the last two studies (See Fig. 3). They developed a Bayes classifier (i.e. a decision Model) to predict molecular sub-type and clinical outcome. It was trained and validated on the Rosenwald Dataset that used the lymphochip platform. The classifier was then independently validated on the Dataset produced by the Shipp group, again using sequence annotations to reconcile the cDNA sequences with the oligonucleotide sequences. This seems to support the biological hypothesis that the “two molecular subtypes” in DLBCL correlate with different biological and clinical behavior. The semantics of the relationship between this Model and these two Datasets is reflected through the visual description and organization in this figure.
On the other hand, the more recent paper by Li et al. (Li, 2006) describes a study that develops and evaluates a specific data-analysis method (i.e. Algorithm) (See Fig. 4). This Algorithm, “Principle Component Analysis and Sliced Inverse Regression”, was applied to both the Rosenwald and Shipp Datasets, as well as to a Dataset produced by a Monte Carlo Simulation. Decision Models were generated and they were validated on an independent subset obtained through one split of the data (148 training samples, 74 training samples). This figure focuses on one algorithm in this Context and relates all the objects (and relationships) that are relevant to the evaluation of this Algorithm.
Model: Object relationships and quality filters
These examples demonstrate that the figures and their underlying complex semantics can not be conveyed by simple retrieval and enumeration of objects returned by Context, i.e. as in the left side of Figure 1. A potentially large number of returned objects need to be organized and displayed intuitively. One aspect of object organization relates to the relationships between the different object types. Such relationships were indicated by edges in the figures. For example, a Paper can describe how an Algorithm is used to Analyze a Dataset. A Model is Produced by running an Algorithm on a Dataset. Models are Validated using more than one Dataset. Grouping objects in annotated relationships can be leveraged in post-retrieval organization and display to provide semantic information about the objects.
All the predictive Models mentioned above underwent some form of validation, expressed via the Validate relationships in the respective figures. The Validate relationship is further specialized via the Validate External and Validate Internal subclasses. Please see the section on evidence annotation in the appendix. As molecular predictive Models mature and get closer to routine clinical practice, it is important to consider the evidence supporting their validity and generalizability. As described by Pepe et al. (Pepe et al. 2001), clinical bioinformatics predictive models typically go through multiple stages of validation before being accepted in standard practice. Therefore, our envisioned system will need to filter different objects based on the strength of supporting evidence. For example, these query results can be narrowed to include only high quality models by appending the following requirements to the query “[get models that …], have been developed using datasets with sample size (n) larger than 200 patients, and that have been validated using an independent dataset.”
The concepts mentioned so far that will support the information retrieval model are described in more detail in the appendix. Now we can revisit Figure 1 in its entirety. It gives an overview of how a query is intended to be processed: A query sets the desired object types, specifies a partial or complete Context(s), and sets conditions for quality filtration. The process is decomposed into three steps: (1) returning objects that are indexed by Context tuples that match the query's Context, (2) filtering out objects based on quality of evidence, and (3) selecting smaller sets of objects by the user and organization of these objects along with their relationships in an intuitive way.
Proof of concept: Molecular prognostic test for breast cancer—MammaPrint®
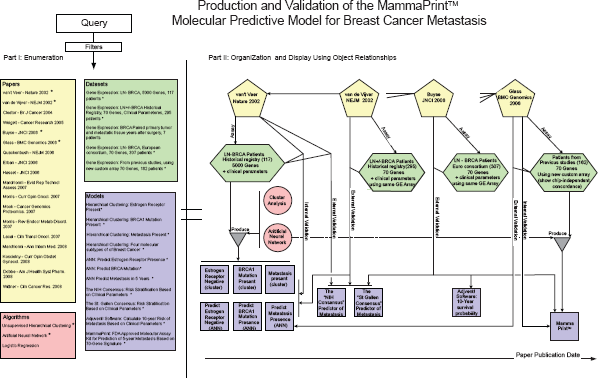
The same semantic representation and organizational principles of Papers, Datasets, Algorithms, and Models that relate to MammaPrint®, the first commercial Breast Cancer molecular prognostic test, are shown in Figure 5 and explained below.
Researchers in the Netherlands (van't Veer and others 2002) analyzed historical breast cancer tissues using a 25,000 sequence oligonucleotide microarray. Seventy genes were found to be predictive of 5-year metastasis in Lymph Node (LN)-negative female patients under the age 55. Unsupervised hierarchical clustering (Algorithm) distinguished the following three characteristics: Estrogen-receptor negative (i.e. can not be treated with the drug Tamoxifen), having BRCA1 germline mutation, and metastasis within 5 years. In other words, three Models were Produced using the hierarchical clustering Algorithm. A supervised machine learning method, Artificial Neural Network (ANN, another Algorithm), was used to construct a classifier (Model), using a “70-gene signature”, that predicts these characteristics. This predictive Model was Validated Internally using a leave-one-out approach. The researchers also showed that this molecular predictive Model was an independent predictor of metastasis from other well-known decision Models that relied solely on clinical parameters (the NIH Consensus and the St. Gallen Consensus). In that paper, not only did the molecular decision Model improve clinical outcome prediction, but it also predicted the same number of patients who had metastasis with fewer false positives. This is important given the morbidity and economic costs associated with adjuvant chemotherapy (Erban and Lau, 2006; Hassett et al. 2006). The 70-“gene signature” Model was Externally Validated (van de Vijver et al. 2002) using 295 consecutive historical patients in a Dataset that is different from the Dataset that was used to Produce that signature. It also provided (Weigelt et al. 2005) the correct decision outcome, i.e. Externally Validated, on primary tumor tissue from 7 patients and on matched metastatic tissue obtained years later from the same patients (not shown in Fig. 5). This validation was not of a clinical, but of a biological hypothesis that: molecular subtype determines the metastatic potential early in the disease as opposed to invasiveness resulting from cumulative mutations.2
That same study Validated a decision Model described elsewhere (also not shown in Fig. 5) that used unsupervised clustering to separate Breast Cancer samples into four molecular subtypes. All matched primary tumors and metastatic tissue belonged to the same molecular subtype.
This figure depicts objects and object relationships that span the development and evolution of the MammaPrint™Model from its earlier versions. The figure also represents the validation of MammaPrint™ across multiple Datasets and its comparison to other Models. Notice that the other clinical predictive models are classical models that do not incorporate molecular data. The information retrieval framework will incorporate classical (non-molecular) clinical predictive Models only when they are relevant to the validation of molecular prediction Models. Otherwise classical Models will not be indexed or stored. Similar to the process described in Figure 1, a query to this domain will return a raw set of objects (Part I, left side). A subset of the raw result may be selected for visual organization and display (right side) of the objects and their relationships (Part II, right side). The detailed prose description of this scenario is presented in the subsection “Proof of Concept: Molecular Prognostic Test for Breast Cancer—MammaPrint®”.
A spin-off commercial company, Agendia™, developed a custom kit that measured gene expression and contained a similar 70-“gene signature” Model, now called MammaPrint®. MammaPrint® was also Produced using the ANN Algorithm and Internally Validated (Glas et al. 2006). The new platform was shown to be concordant with the previous 25,000 oligonucleotide chip (Glas and others 2006) (thus Externally Validating that Dataset's corresponding Model). MammaPrint® was Externally Validated through multi-center European consortium study (Buyse et al. 2006). It was also compared to known clinical decision Models, including one based on a software, Adjuvant!, that calculates 10-year survival probability based on clinical parameters.
Discussion and Future Work
Some public resources currently implement some but not all aspects of our intended functionality and not in an integrated retrieval framework as was discussed in this paper. For example, PharmGKB's clinical outcomes are restricted to outcomes of therapy, and exclude diagnostic and prognostic markers. Oncomine's representation and organization of oncology molecular datasets does not cover decision Models, the original Algorithms by which these models were produced, or their validation methods. Datasets and Papers are MeSH-indexed in GEO/PubMed, but their relationships to respective Models, Algorithms, and Contexts are not explicit. The proposed framework is designed to compliment existing resources and extend current representations to cover molecular clinical predictive models and their related modalities. Our choice to model this domain using an OWL ontology was made with the goal of semantic integration of this framework with existing knowledge sources. Whenever possible we associate objects in our database with their counterparts in external databases, e.g. using PubMed uid for papers and GEO accession numbers for datasets.
Most existing clinical predictive models do not incorporate molecular features. Classical predictive models that are purely based on clinical parameters are outside the scope of this information retrieval framework; however, classical models will be incorporated only when they exist within the context of molecular predictive models. For example, we did include the International Prognostic Index model in the DLBCL case study, and the St. Gallen Consensus model in the MammaPrint™ validation case study. Similarly, storing and annotating gene signatures that predict underlying biological behavior without clinical outcomes is outside the scope of this framework. Again, some molecular clinical predictive models incorporate aspects of purely biological signatures, so we will also include those only when they exist within the context of clinical models. For example, the early DLBCL models (Fig. 2) that identified the underlying biological behavior of DLBCL (as APB-like or GC-like) did correlate with clinical outcomes and therefore they were included in the framework. Using molecular signatures that measure (EGF-R) receptor activity for choice of treatment with tyrosine kinase inhibiting drugs is another example (not discussed in this paper) that comes to mind of what will be included in this framework.
The focus of the present paper is the underlying information retrieval model and not the system's implementation and inference mechanisms which will be described elsewhere (please see Appendix). When developing the formalisms described in this paper, we deliberately selected the minimal set of classes and properties that is expressive enough to allow for semantic organization of the domain. This level of simplicity is intended to enable automated methods for building the knowledgebase. Our current research is focused on building and validating machine learning models that can correctly annotate the Contexts described in clinical bioinformatics papers, and that can also correctly identify the validation methods that are employed in those papers.
Conclusion
While clinically-oriented research exploring gene expression microarrays, mass spectrometry, SNP arrays and other high-throughput molecular assays has followed an exponential growth in recent years, to date there is no general purpose system that allows researchers and clinicians to find models, papers, data, and other related information in this emerging field using a unified and friendly interface. In the present paper we propose a framework for such interface and demonstrate the complexity of its required functionality. Our long-term goal is to construct a system that addresses this need. As a significant first step, we developed a formalism that supports storage and retrieval of a multiplicity of clinical bioinformatics objects such as published papers, datasets, decision models, and discovery and inference algorithms. This formalism opens the way for automated methods that support the knowledgebase's creation and annotation. In addition, it allows for a second layer of organization of objects returned by queries based on their (1) interrelationships and (2) strength of methodological validation. We demonstrated the power of this model in the complicated domain of diffuse large B-cell lymphoma. In future work we plan to deploy and test a prototype system based on the model of the present paper applied to biomarker discovery for other malignancies.
Footnotes
Appendix
Disclosure
The authors report no conflicts of interest.
References
1.
AitkenJ.S., WebberB.L. and BardJ.B.2004. Part-of relations in anatomy ontologies: a proposal for RDFS and OWL formalisations.Pac. Symp. Biocomput., 166–77.
2.
AlizadehA.A., EisenM.B., DavisR.E.2000. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling.Nature, 403(6769): 503–11.
3.
AltmanR.B., FlockhartD.A., SherryS.T.2003. Indexing pharmacogenetic knowledge on the World Wide Web.Pharmacogenetics, 13(1): 3–5.
4.
AndoY., SakaH., AndoM.2000. Polymorphisms of UDP-glucuro-nosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis.Cancer Res., 60(24): 6921–6.
5.
AphinyanaphongsY., TsamardinosI., StatnikovA.2005. Text Categorization Models for High-Quality Article Retrieval in Internal Medicine.Journal of the American Medical Informatics Association, 12(2): 207–16.
6.
AshburnerM., BallC.A., BlakeJ.A.2000. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium.Nat. Genet., 25(1): 25–9.
7.
BaaderF.2003. The description logic handbook theory, implementation, and applicationsCambridge University Press, Cambridge, UK.
8.
BallC.A., BrazmaA., CaustonH.2004. Submission of microarray data to public repositories.PLoS. Biol., 2(9): E317.
9.
BarrettT., TroupD.B., WilhiteS.E.2007. NCBI GEO: mining tens of millions of expression profiles—database and tools update.Nucleic Acids Res., 35 Database issue: D760–D65.
10.
BellD.W., LynchT.J., HaserlatS.M.2005. Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: molecular analysis of the IDEAL/INTACT gefitinib trials.J. Clin. Oncol., 23(31): 8081–92.
11.
BrazmaA., HingampP., QuackenbushJ.2001. Minimum information about a microarray experiment (MIAME)-toward standards for microarray data.Nat. Genet., 29(4): 365–71.
BurgunA.2006. Desiderata for domain reference ontologies in biomedicine.J. Biomed. Inform., 39(3): 307–13.
15.
BuyseM., LoiS., van't VeerL.2006. Validation and Clinical Utility of a 70-Gene Prognostic Signature for Women With Node-Negative Breast Cancer.jnci., 98(17): 1183–92.
16.
CiottiM., ChenF., RubaltelliF.F.1998. Coding defect and a TATA box mutation at the bilirubin UDP-glucuronosyltransferase gene cause Crigler-Najjar type I disease.Biochim. Biophys. Acta., 1407(1): 40–50.
17.
CouzinJ.2007. Diagnostics. Amid debate, gene-based cancer test approved.Science, 315(5814): 924.
GlasA.M., FlooreA., DelahayeLJMJ.2006. Converting a breast cancer microarray signature into a high-throughput diagnostic test.BMC Genomics, 7: 278.
23.
GolubT.R., SlonimD.K., TamayoP.1999. Molecular Classification of Cancer: Class Discovery and Class Prediction by Gene Expression Monitoring.Science, 286(5439): 531–7.
24.
HassettM.J., O'MalleyA.J., PakesJ.R.2006. Frequency and Cost of Chemotherapy-Related Serious Adverse Effects in a Population Sample of Women With Breast Cancer.jnci., 98(16): 1108–17.
25.
HoffmanM., ArnoldiC. and ChuangI.2005. The clinical bioinformatics ontology: a curated semantic network utilizing RefSeq information.Pac. Symp. Biocomput., 139–50.
26.
InnocentiF., UndeviaS.D., IyerL.2004. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan.J. Clin. Oncol., 22(8): 1382–8.
27.
JonesA., HuntE., WastlingJ.M.2004. An object model and database for functional genomics.Bioinformatics, 20(10): 1583–90.
KnublauchH., MusenM.A. and RectorA.L.2004. Editing description logic ontologies with the protege-owl plugin.International Workshop on Description Logics..
30.
LiL.2006. Survival prediction of diffuse large-B.-cell lymphoma based on both clinical and gene expression information.Bioinformatics, 22(4): 466–71.
31.
LynchT.J., BellD.W., SordellaR.2004. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib.N. Engl. J. Med., 350(21): 2129–39.
32.
ManduchiE., GrantG.R., HeH.2004. RAD and the RAD Study-Annotator: an approach to collection, organization and exchange of all relevant information for high-throughput gene expression studies.Bioinformatics, 20(4): 452–9.
33.
MarshS. and McLeodH.L.2006. Pharmacogenomics: from bedside to clinical practice.Hum. Mol. Genet., 15(1): R.89–R.93.
34.
MathewJ.P., TaylorB.S., BaderG.D.2007. From Bytes to Bedside: Data Integration and Computational Biology for Translational Cancer Research.PLoS Computational Biology, 3(2): e12.
35.
McGuinnessD.L., and van HarmelenF.2004. OWL Web Ontology Language Overview.W3C Recommendation, 10: 2004–03.
NtzaniE.E. and IoannidisJ.P.2003. Predictive ability of DNA microarrays for cancer outcomes and correlates: an empirical assessment.Lancet, 362(9394): 1439–44.
38.
OliverD.E., RubinD.L., StuartJ.M.2002. Ontology development for a pharmacogenetics knowledge base.Pac. Symp. Biocomput., 65–76.
39.
onso-CalvoR., MaojoV., BillhardtH.2007. An agent- and ontology-based system for integrating public gene, protein, and disease databases.J. Biomed. Inform., 40(1): 17–29.
40.
PepeM.S., EtzioniR., FengZ.2001. Phases of biomarker development for early detection of cancer.J. Natl. Cancer Inst., 93(14): 1054–61.
41.
Perez-ReyD., MaojoV., Garcia-RemesalM.2006. ONTOFUSION: ontology-based integration of genomic and clinical databases.Com-put. Biol. Med., 36(7-8): 712–30.
42.
QuackenbushJ.2006. Microarray analysis and tumor classification.N. Engl. J. Med., 354(23): 2463–72.
43.
RhodesD.R., Kalyana-SundaramS., MahavisnoV.2007. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles.Neoplasia, 9(2): 166–80.
44.
RhodesD.R., YuJ., ShankerK.2004. ONCOMINE: a cancer microarray database and integrated data-mining platform.Neoplasia, 6(1): 1–6.
45.
RosenwaldA., WrightG., ChanW.C.2002. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma.N. Engl. J. Med., 346(25): 1937–47.
46.
RossJ.S. and GinsburgG.S.2003. The integration of molecular diagnostics with therapeutics. Implications for drug development and pathology practice.Am. J. Clin. Pathol., 119(1): 26–36.
ShippM.A., RossK.N., TamayoP.2002. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning.Nat. Med., 8(1): 68–74.
49.
SimonR.2005. Roadmap for developing and validating therapeutically relevant genomic classifiers.J. Clin. Oncol., 23(29): 7332–41.
50.
SimonR. and ZhaoY.BRB.-ArrayTools Data Archive for Human Cancer Gene Expression: A Unique and Efficient Data Sharing Resource.Cancer Informatics6, 9–15. 4–21–2008. Ref Type: Journal (Full).
51.
SioutosN., deC.S., HaberM.W.2007. NCI Thesaurus: a semantic model integrating cancer-related clinical and molecular information.J. Biomed. Inform., 40(1): 30–43.
52.
SmithB., CeustersW., KlaggesB.2005. Relations in biomedical ontologies.Genome Biol., 6(5): R.46.
53.
SobieE.A., GuatimosimS., SongL.S.2003. The challenge of molecular medicine: complexity versus Occam's razor.J. Clin. Invest., 111(6): 801–3.
54.
The International Non-Hodgkin's Lymphoma Prognostic Factors Project1993, “A predictive model for aggressive non-Hodgkin's lymphoma. The International Non-Hodgkin's Lymphoma Prognostic Factors Project.”N. Engl. J. Med., 329(14): 987–94.
55.
van de VijverM.J., HeY.D., van't VeerL.J.2002. A Gene-Expression Signature as a Predictor of Survival in Breast Cancer.New England Journal of Medicine, 347(25): 1999.
56.
van't VeerL.J., DaiH., van de VijverM.J.2002. Gene expression profiling predicts clinical outcome of breast cancer.Nature, 415(6871): 530–6.
57.
VoseJ.M.1998. Current approaches to the management of non-Hodgkin's lymphoma.Semin. Oncol., 25(4): 483–91.
58.
WeigeltB., HuZ., HeX.2005. Molecular Portraits and 70-Gene Prognosis Signature Are Preserved throughout the Metastatic Process of Breast Cancer.Cancer Research, 65(20): 9155–8.
59.
WilczynskiN., MorganD., HaynesR.B.2005. An overview of the design and methods for retrieving high-quality studies for clinical care.BMC Medical Informatics and Decision Making, 5(1): 20.
60.
WrightG., TanB., RosenwaldA.2003. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B-cell lymphoma.Proc. Natl. Acad. Sci. U.S.A, 100(17): 9991–6.