
Abstract
Integrative cancer biology research relies on a variety of data-driven computational modeling and simulation methods and techniques geared towards gaining new insights into the complexity of biological processes that are of critical importance for cancer research. These include the dynamics of gene-protein interaction networks, the percolation of sub-cellular perturbations across scales and the impact they may have on tumorigenesis in both experiments and clinics. Such innovative ‘systems’ research will greatly benefit from enabling Information Technology that is currently under development, including an online collaborative environment, a Semantic Web based computing platform that hosts data and model repositories as well as high-performance computing access. Here, we present one of the National Cancer Institute's recently established Integrative Cancer Biology Programs, i.e. the Center for the Development of a Virtual Tumor, CViT, which is charged with building a cancer modeling community, developing the aforementioned enabling technologies and fostering multi-scale cancer modeling and simulation.
Keywords
Background
The completion of the Human Genome Project catalyzed a systems view of biomedicine which may well have a dramatic impact on 21st century's life sciences all together (e.g. Deisboeck T.S. and Kresh J.Y., 2006). The concept, attributed to Aristotle, that “the whole is more than the sum of its parts” suggests that dissecting an organism, tissue or cell into ever smaller monomers and hoping to be able to piece it back together afterwards will not work if the underlying mechanisms are anything but linear and involve dynamic relationships. While systems theory per se is hardly new (see e.g. seminal work by Bertalanffy v. L., 1968) based on some pioneering yet rather theoretical studies on complex systems in biology (e.g. Kauffman S.A., 1993) the notion of systems biology as the research approach that integrates biology, medicine, computation and technology to comprehend biological information processing has recently been embraced also by mainstream science (e.g. Ideker et al. 2001; Kitano, 2002). Consequently, this has led to a wave of academic, corporate and governmental efforts in this emerging field. Already at this nascent stage it has become abundantly clear, however, that the transition from classic reductionism driven biomedical science to a systems level understanding of biological processes requires more than access to high-performance computing only. Rather, it needs a new understanding of how multi-scaled biological systems have to be investigated with an approach that integrates computation and experiment and, ultimately, in the case of disease processes, how they can be diagnosed and treated—as systems.
If a tumor is thought of as a dynamic self-organizing biosystem, one can argue that cancer is an almost ideal case to apply the considerable strengths of this new systems biology concept. This is not only because we are still far from deciphering the complexity of all the factors involved in tumorigenesis but also since the countless experimental and clinical studies devoted to it continue to generate an ever growing amount of disparate data with little chance of connecting the ‘dots’ using conventional scientific approaches only. There is no doubt then that innovative computational modeling and simulation, in conjunction with appropriately designed experiments, will rapidly become a valuable if not crucial tool for this new scientific path in cancer biology. Specifically, cutting edge multi-scale computational modeling will be able (a) to help generate experimentally testable hypotheses, (b) to integrate diverse data, and ultimately, (c) to predict outcome also for clinical purposes. Currently, mechanistic dynamical simulations and inferential data mining constitute the two main approaches in interdisciplinary cancer systems biology research with significant progress. (1) For instance, molecular pathway simulation has shown promise exemplified through the work by Araujo et al. (2005) who have developed a mathematical model to investigate combination therapy with kinase inhibitors by building upon theoretical studies of the epidermal growth factor receptor (EGFR) pathway (Kholodenko et al. 1999). Another example is Athale et al. (2005, 2006) who, based on previous works by Mansury and Deisboeck (2003, 2004), have modeled a proposed cellular phenotypic switching mechanism also in the EGFR signaling pathway. Most recently, Zhang et al. (2007) have then extended this work in order to simulate the dynamics of EGFR gene-protein interaction profiles, alternating cell phenotypes and emergent multi-cellular patterns with a three dimensional agent-based multi-scaled model (Figure 1). (2) On the other hand, because it is now possible to extract knowledge from large-scale data sets employing advanced data mining techniques (Khalil and Hill, 2005), progress has been made in detecting patterns and correlations in the data that lead to new hypotheses about possible interactions such as on the protein-protein and gene-protein level (e.g. Yeger-Lotem et al. 2004). It is thus a reasonable goal to combine these two promising paths in the future.

Nonetheless, despite, or more accurately, precisely because of the significant progress made, several challenges for the field have become apparent as well. That is, the absence of a cancer modeling dedicated, collaborative community or network has led to a lack of shared standards or even guidelines let alone unifying platforms to archive, exchange and integrate the many distinct computational and mathematical models that have been and are being developed. This inevitably led to redundancy in some cases where multiple models on for instance ‘cell migration’ have been developed over the years whereas other tumor characteristics have received far less attention. Overall, the result is a diminished impact of the modeling field on experimental cancer research and a loss of potentially valuable time for clinical studies. Therefore, particularly for projects that exceed the capacities of a single team, access to distributed scientific and technical expertise, exchange of knowledge and sharing of biomedical data, modeling algorithms and analysis tools appear to be most critical.
To address these issues and to advance the field of cancer systems biology, the National Cancer Institute, NCI, has recently established the Integrative Cancer Biology Program, ICBP (URL: http://icbp.nci.nih.gov/). One of these nine ICBPs is the
Introducing CViT
CViT's mission is threefold: (1) to establish a
Building a Cancer Modeling Community: ‘CViT.org’
Cancer research has always been an international enterprise with pockets of critical expertise being developed at numerous sites all over the world and thus now, more so than ever, large-scale consortium projects have to go beyond institutional boundaries to accomplish a set task that would otherwise exceed the resources available at a single site. Given the fact that this entails setup and management of long-distance collaborations that also cross multiple disciplines, a new more flexible collaborative environment has to be created. CViT has built such a user-friendly online platform, CViT. org (URL: http://www.cvit.org) that employs a username & password-protected ‘wiki’-type environment to post and discuss content (Figure 2, left). A relatively large portion of the regularly updated information such as researcher profiles (Figure 2, right), resources, tutorials and software tools, is already publicly-accessible as part of CViT's commitment to community outreach. For the continuously growing CViT group of investigators which includes scientists from dozens of institutions around the world, CViT.org provides its participating investigators a number of advanced tools to rapidly communicate, thus facilitating dissemination of knowledge and fostering collaborations: (1) CViT.org offers

CViT Home Page (left) and Investigator Profiles (right).
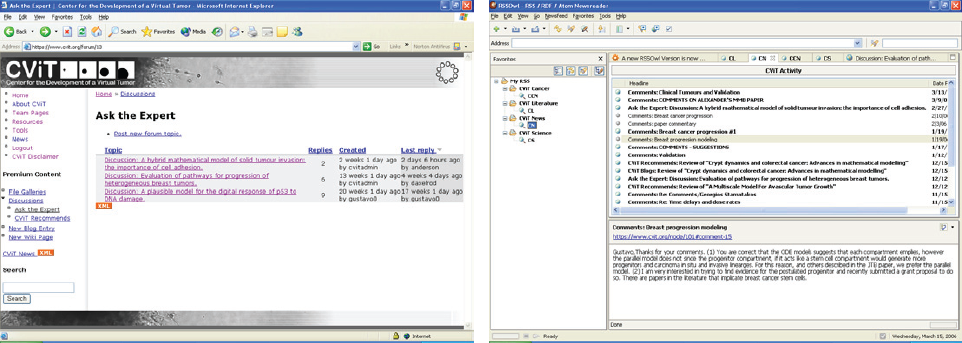
CViT Blog (left) and RSS Feeds (right).

CViT Annotation System.
Knowledge Integrated Modeling and Semantic Layered Research Platform Prototype
CViT's Digital Model Repository (below) will itself be part of a wider application system currently named
Berners-Lee,T., Hendler, J., Lassila, O. (2001) The Semantic Web, Scientific American, May 2001 (URL: http://www.scientificamerican.com/article.cfm?articleID=00048144-10D2-1C70-84A9809EC588EF21&catID=2).
Miller E, Swick R, Brickley D (2003) ‘W3C Resource Description Framework (RDF)’ (URL: http://www.w3c.org/RDF/).
EBI, I3C, IBM (2003) ‘Life Science Identifiers RFP Response Revised Joint Submission’, Object Management Group Document lifesci/03-10-01 (URL: http://www.omg.org/cgi-bin/doc?lifesci/2003-10-01).
The
CViT researchers using the KIM application will be provided with a “Biologists Workbench” application program that can be customized to aid in the tasks that scientist carries out in his/her day to day work and collaborations with colleagues. This application will be based on the
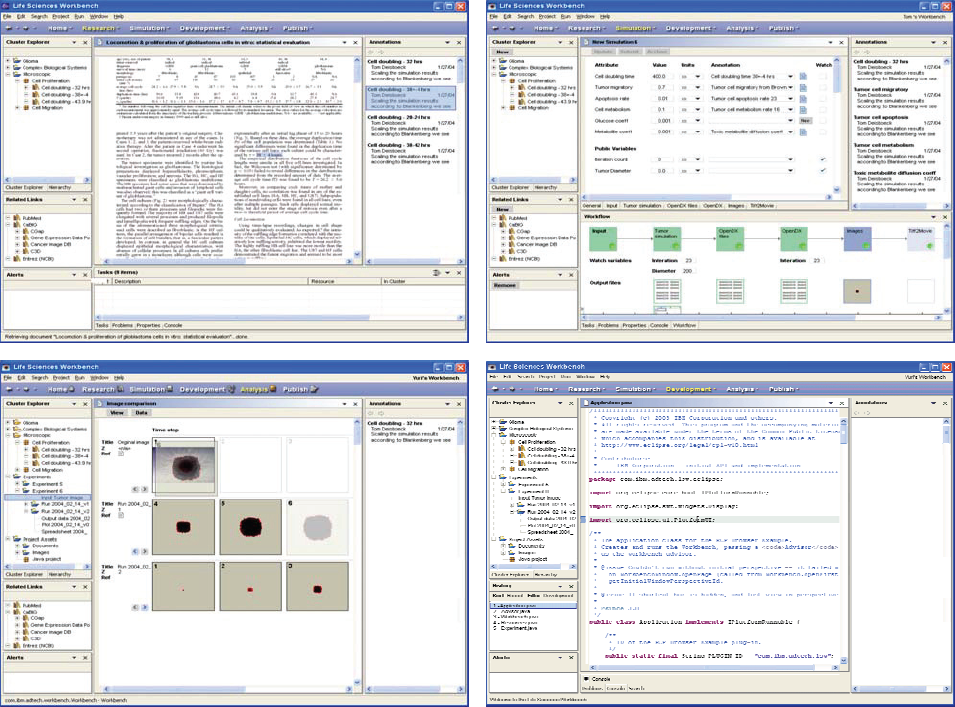
Knowledge Integrated Modeling. Employing SLRP annotated data (e.g. from PubMed listed manuscripts; top left) are captured and used as model input parameters (top right). Deploying the code (language editing perspective, bottom right) to a high-performance compute environment yields in silico results that can then be compared with experimental data such as the microscopy images of tumors cultured in vitro (bottom left), or back to data published in the literature. This feedback allows continuous refinement of the (DMR-archived) algorithm(s) and spurs design and development of experiments.
The Digital Model Repository (DMR)
The Digital Model Repository will utilize the
We note in this context that any model description consists not only of the algorithm, its code implementation and, if available, its markup language representation (see below), but also of, for instance, related experimental data, simulation results, visualizations and manuscripts (Figure 6). Researchers using the SLRP system to track their modeling will automatically create and store semantic links to the inputs and output of any particular model run, along with LSID references to the actual source code modules that were executed for any particular simulation execution run. The system will automatically document and make directly accessible the knowledge required to exactly reproduce a digital experiment without fear that the an incorrect version of the source code or input parameter data is being used.

Usage of CViT's DMR content by participating investigators and their institutions will be enabled through an already posted open source license that is approved by the National Cancer Institute and in compliance with guidelines put forward by the Cancer Biomedical Informatics Grid, caBIG. Close interaction with caBIG is planned and will likely include caBIG standards compliant web-service to both use and provide data and access to high-performance computing as well as specialized analysis algorithms. Finally, more work may be required at a later stage on the representation level if the models in CViT's repository are to be integrated with those in other non-cancer focused digital repositories. Examples for such model representations include CellML (URL: http://www.cellml.org/; its repository can be accessed at URL: http://www.cellml.org/models) which has already found use in the larger biomedical modeling community. Since CellML includes MathML, equations are straightforward to add. Additionally, the RDF section of CellML appears to be adaptable to describe other model parameters and would potentially integrate well with CViT's overall underlying system platform that is based on RDF as mentioned earlier. Another alternative is SBML (URL:http://sbml.org/index.psp; used e.g. by the repository of the European Bioinformatics Institute (URL:http://www.ebi.ac.uk/biomodels/)), which, although focused primarily on the biochemical pathway level, has however more libraries and parsers available and has been implemented in many biological modeling tools.
Conclusion
The new paradigm of systems biology holds great promise for progress, from biomedical basic science (e.g. Kitano, 2002) to drug discovery (e.g. Hood and Perlmutter, 2004), and therefore in particular for cancer research (Coffey, 1998; Hornberg et al. 2006; Khalil and Hill, 2005; Waliszewski et al. 1998). Integrative cancer systems biology is built on the premise that a better understanding of the complexity of tumorigenesis requires design, development and implementation of novel, data-driven in silico models that account for the multi-scaled processes involved. However, cancer modeling algorithms are usually developed in a non-standardized fashion, often very specific in addressing a particular problem of interest, thus commonly lack widespread distribution, therefore acceptance in and feedback from experimentalists and clinicians. While one must concede that currently there is no perfect cancer model available or in sight, this fact all the more emphasizes the need of sharing and comparing available models as well as the data that went into them or are derived from them, thus for archiving algorithms just as much as for exchanging concepts and results. It is here where CViT will make a difference. Its growing international group of investigators not only represents already an unparalleled level of expertise, it also allows for rapid dissemination of information relevant to the field using the tools made available at CViT.org. The technologies CViT currently develops, specifically its semantic layered research platform with its digital model repository and knowledge integrated modeling workflow address critical needs of the community and thus undoubtedly will help advance the field of integrative cancer biology also beyond the current scope of NCI's ICB program. All this creates a cutting edge environment that can make CViT's long term vision for multi-scale cancer systems biology research a reality: i.e. the development a module-based cancer modeling tool-kit that can be specified as need be in support of personalized medicine.
Footnotes
Acknowledgments
This work has been supported by NIH grant CA 113004 and by the Harvard-MIT (HST) Athinoula A. Martinos Center for Biomedical Imaging and the Department of Radiology at Massachusetts General Hospital.