
Abstract
Gene expression profiling provides tremendous information to help unravel the complexity of cancer. The selection of the most informative genes from huge noise for cancer classification has taken centre stage, along with predicting the function of such identified genes and the construction of direct gene regulatory networks at different system levels with a tuneable parameter. A new study by Wang and Gotoh described a novel Variable Precision Rough Sets-rooted robust soft computing method to successfully address these problems and has yielded some new insights. The significance of this progress and its perspectives will be discussed in this article.
Keywords
Gene expression profiles (GEP) either by microarray or by Serial Analysis of Gene Expression (SAGE) provide us with data of unparalleled wealth, but cancer as a system failure is still mysterious. Many existing methods utilize too many genes to obtain discriminative features associated with cancer, and are unclear or not interpretable at a biological level. Developing simpler rule-based models with as few marker genes as possible is preferable. Ideally, such hub genes could naturally exhibit biological relevance. But good research is never simple and requires hard work: there is no “free lunch” researchers. However, based on a Variable Precision Rough Set (VPRS) core
1
with the introduction of
A Brief Introduction of the α Depended Degree Rough Set Soft Computing Approach
Firstly, rough set theory neds to be understood. In this theory, f/is a universe of discourse and

where
If
For some datasets, it is difficult to detect the discriminative features based on the canonical depended degree because of its excessively rigid definition. Therefore, Wang and Gotoh introduced
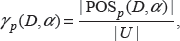
where
Wang and Gotoh created classifiers based on decision rules. One decision rule in the form of “
For each determined α value, only the genes with
User-Friendly Theory, Practical Simplicity and Biological Interpretability
Biologists generally speak different “languages” from mathematicians. Unlike statistical methods, this novel method, the Bimodality Index, 7 sought to be interpretable for biological relevance simple for cancer classification in both theory and practice. Importantly, this method allows a straightforward inference of the direct gene regulatory network. All the gene selection, classification and network construction processes in this method correlate with well biologically meaningful decision rules, such as tumor vs. normal cells, up- vs. down-regulation, and positive vs. negative regulation. This contrasts with the process of many other methods, where the classifying power of the gene expression level and the biological importance of that gene are generally only weakly related and thus many biomarker candidates could turn out to be false positives.
This novel method is rooted on the rough sets theory (RS) seminally proposed by Pawlak
8
for analysis of inconsistent, incomplete, imprecise and precise data. The main advantage of RS is that it does not need any preliminary or additional information about data, e.g. probability in statistics or basic probability assignment in Dempster–Shafer theory. RS has been successfully applied in the areas of medicine and pharmacology.
9
Its application in cancer classification and prediction has begun.2–5 As the inhibition of a single molecular target can alter the morphology of tumor cells in lrECM and reduce tumor growth
This theory itself may be akin to our routine identification (or classification) of objects in the real-world setting. The rationale is first to filter lots of redundant information (i.e. noise) but to retain the critical information (i.e. signal). This is followed by making decision rules based on core information and classifying the whole dataset. In order to extract the hidden meaningful rules, we sometimes need to lose some rigid definitions. Thus Wang and Gotoh introduce the flexible α depended degree under soft computing consideration. This allows some single genes or gene pairs to have strong class discriminatory power, although they would be ignored with the conventional attribute depended degree. 2 Interestingly, this also enables us to infer the networks and modules.
In fact, Wang and Gotoh reject the attribute reductions in classic rough set theory due to its high com putational expense, uncertainty of predictive performance and non-uniqueness. 2 Because of depended degree, they use the entropy-based discretization method 12 for discrete gene expression values within datasets.2–5 The stopping point of the recursive step for this algorithm depends on the minimum description length (MDL) principle and the discretization was implemented in the Waikato Environment for Knowledge Analysis (WEKA) package, 13 which gives open access to a collection of state-of the-art techniques in machine learning algorithms for data mining tasks; these algorithms can either be applied directly to a dataset or called from user's own Java code, so it is an excellent unified “workbench” not only for data pre-processing, classification, regression, clustering, association rules and visualization but is also well suited for developing new machine learning schemes.
This process is more or less streamlined. In the discretized decision table, Wang and Gotoh found that most genes were unable to distinguish different classes and were removable, while some genes can distinguish different classes by decision rules. 5 They achieved very high leave-one-out-cross-validation (LOOCV) accuracy for an array of datasets.2–5 The reported accuracy is superior to or comparable with other established approaches.2–5
In their new work on the colon cancer dataset, Wand and Gotoh identified 18 discriminative hub genes for cancer. Ten of these (e.g.
Inference of the Gene Regulatory Network
Obtaining a direct regulatory network of these discriminative hub genes is of particular interest. Functional entities, such as pathways nad signalling networks are more robust descriptors than gene lists. 15 The similarity measures, such as Pearson's correlation and mutual information 16 cannot characterize the cause–effect gene regulatory relations in undirected networks very well. In contrast, directed gene regulatory networks, such as Bayesian networks, Boolean networks, Ordinary Differential Equations or IDA.17,18 can explore the cause–effect regulatory relations and provide better insights into biological systems than the co-expression relation. Moreover, most previous efforts utilized all gene expression data from microarrays so that the authentic gene interactions were covert due to many genes that were unrelated to cancer. However, it is expected that a few highly class-discriminative hub genes could greatly enhance the authenticity and confidence of computed gene interaction networks.
Following the identification of hub genes, Wang and Gotoh investigated the gene regulatory network by employing the method described above. The details of this method are as follows: one gene instead of a class is used as the decision attribute. If “GENE-I” is substituted for “Class label” in a decision table, GENE-I is regarded as the decision attribute with two distinct values: up-regulation and down-regulation, and a new derivative table can be obtained. Likewise, Wang and Gotoh implement the discretization of this derivative table to obtain another newly derived table. Applying the same learning algorithm to this latest derived table, they can induce the decision rules linking GENE-I to GENE-II: if the expression level of GENE-I in one sample is not greater than value A, then GENE-II is down-regulated; otherwise, GENE-II is up-regulated. In other words, if GENE-I is down-regulated, then Gene-II is down-regulated; if Gene-I is up-regulated, then Gene-II is up-regulated. They are not necessarily true in reverse. Therefore, a directed regulatory relation of GENE-I to GENE-II, a positive one, is established. 5
Similarly, Wang and Gotoh regard each of the 18 identified genes as the decision attribute in turn, and examine the regulatory relations that the other genes exert on them. They constructed all their network graphs using Cytoscape software. 19 They analyzed one network containing only these 18 genes, and another containing genes other than these 18. The first networke one orchestrates the core of the latter in the genome. Modules constitute the ‘'building blocks” of molecular networks. To explore the modularity of networks, Wang and Gotoh use the Cytoscape plugin MCODE 19 to analyze the network constructed and detected two significant modules, one of which forms a feed-forward loop. They conclude that the co-regulation of multiple activators could be at least partly responsible for the occurrence of tumors. Further, they chose the Cytoscape plugin BiNGO 20 to perform a Gene Ontology (GO) based enrichment analysis of the two modules. Other gene functional analysis, such as Gene Set Enrichment Analysis (GSEA), could also be useful. Finally, they observed that in colon cancer, the gene regulatory network, the up-regulated genes are regulated by more genes than down-regulated ones, while the down-regulated genes regulate more genes than up-regulated ones; secondly, tumor suppressors inhibit tumor activators and activate as many other tumor suppressors as possible. In contrast, tumor activators activate other tumor activators and inhibit as few tumor suppressors as possible. 5 A fascinating question: is it true for other cancers and how about its validation of wet-lab experiments?
This method is a new option for cancer classification and direct gene regulatory network inference. For these processes, it exhibits its inherent biological relevance. Finally, this method out-performs or at least matches other approaches, though LOOCV may have a large variance of accuracy.2–5 Taking into account its other merits, especially its simplicity, this is a great way to explore the cancerogenesis according to Occam's Razor: the simple theory is preferable to the complex one. A scheme of a “free-lunch” toolkit for cancer classification and networks is shown in Figure 1.

Scheme of the “free lunch” toolkit for cancer classification at the network level and beyond. Arrow: executed Dash arrow: being executed “Free lunch” kit codes: the programming codes for cancer classification, hub gene identification and inference of gene regulatory network under GNU GPL.
Future Directions
This kind of cause–effect inference could have practical value in the prioritization and design of perturbation experiments. Of course, only verification via follow-up wet-lab studies rather than published literature could prove that the conclusions from this new study are perfectly valid and reliable, though, theoretically, the process always demonstrates biological relevance, which may have already sparked the curiosity and passion of biologists and clinicians.
In the near future, a wide variety of datasets, such as subtype or multi-class cancer microarray data, microRNA array data, Serial Analysis of Gene Expression (SAGE) data and proteomic data could challenge the “free-lunch” toolkit. Thus far, we have identified seven highly discriminative (hub) genes in the SAGE breast cancer dataset,
21
which has approximately 2.7 million tags and which has 27 samples, each of which are described as lymph node [LN(+)] and [LN(–)] primary breast tumors. All identified genes have high classification accuracy using this method under α = 0.8 (Results are presented in Table 1). These seven hub genes are very interesting and informative for their biological relevance. First, it is well known that the role of the ATF2/AP1 complex and its network is at the hub of tumorigenesis.22,23 and this has been reflected by a high classification accuracy of 88.89%. ATF2 communicates with an array of cell signalling pathways that are important for mammary tumors, e.g. TGFbeta. This emphasizes that comprehensive understanding of how ATF2 functions promises to provide new avenues for therapeutic intervention in breast cancer. CARD10/CARMA3 has a physical and functional interaction with IkappaKgamma-NEMO in lymphoid and non-lymphoid cells, is required for GPCR-induced NF-kappaB activation
24
and is important in LPA-induced cancer cell
The seven hub genes identified in the breast cancer SAGE dataset.

Importantly, the ENCODE project tells us that at least 93% of the analyzed human genome is transcribed in different cells into biologically meaningful RNAs that could greatly exceed the ∼1.2% encoding proteins. 26 More and more attention is being given to RNA, especially Linc RNAs, microRNAs and antisense RNAs. However, the protein levels and IHC staining have a greater variety of available assays in the clinical setting. Archimedes once said, “Give me a lever long enough and a fulcrum on which to place it, and I shall move the world”. Recent advances in deep-sequencing application in ChIP-seq, SAGE-seq, HITS-CLIP 27 and MALDI-TOF mass-spec in proteomics and the exponential increase of available profiling datasets may act as a metaphorical fulcrum. The method of Wang and Gotoh, together with others, e.g. the Bimodality Index.7,29 have made advances in the direction of being the lever. Simple, yet powerful and reliable techniques like the “free-lunch” toolkit could pave the way to unveiling the mystery of cancer.
Another direction is to dissect cancerogenesis
The perturbations of gene regulatory networks could be essentially responsible for cancinogenesis
5
and the therapeutic recovery could reflect the flexibility and robustness of biological system. It will be exciting to perform
Disclosure
This manuscript has been read and approved by the author. This paper is unique and is not under consideration by any other publication and has not been published elsewhere. The author and peer reviewers of this paper report no conflicts of interest. The author confirms that they have permission to reproduce any copyrighted material.
Footnotes
Acknowledgement
The author is deeply grateful to Dr. Wang for programming the code for analysis of SAGE data and for giving his opinion.