
Abstract
Gene expression signatures are commonly used to create cancer prognosis and diagnosis methods, yet only a small number of them are successfully deployed in the clinic since many fail to replicate performance on subsequent validation. A primary reason for this lack of reproducibility is the fact that these signatures attempt to model the highly variable and unstable genomic behavior of cancer. Our group recently introduced gene expression anti-profiles as a robust methodology to derive gene expression signatures based on the observation that while gene expression measurements are highly heterogeneous across tumors of a specific cancer type relative to the normal tissue, their degree of deviation from normal tissue expression in specific genes involved in tissue differentiation is a stable tumor mark that is reproducible across experiments and cancer types. Here we show that constructing gene expression signatures based on variability and the anti-profile approach yields classifiers capable of successfully distinguishing benign growths from cancerous growths based on deviation from normal expression. We then show that this same approach generates stable and reproducible signatures that predict probability of relapse and survival based on tumor gene expression. These results suggest that using the anti-profile framework for the discovery of genomic signatures is an avenue leading to the development of reproducible signatures suitable for adoption in clinical settings.
Background
Despite many advances in cancer treatment, early detection remains the most promising avenue in terms of patient survival.1–5 While there have been many attempts at devising early cancer screening techniques, many approaches remain inefficient in clinical settings and are not pragmatic because of lack of cost-effectiveness or requirement of invasive procedures.6–8 Genomic screening techniques are a promising approach in this area. Continuing advances in high-throughput technology make these approaches both cost- and time-effective. Certain types of genomic data, such as gene expression derived from peripheral blood, are minimally invasive as well.
The main difficulty in developing such techniques has been the lack of stable markers in cancer gene expression profiles. Apart from a few exceptions,9,10 many gene signatures have failed to reproduce their results when tested on independently obtained data, 11 indicating that the signature is not adequately robust to be deployed in a clinical setting.
However, a recent work by Corrada Bravo et al. 12 demonstrates that by modeling consistent increased gene expression variability across cancer types, a statistical model can be developed that provides a stable and robust predictor of cancer that works well across multiple cancer types. The underlying observation behind this approach is that certain genes will consistently show higher across-sample variability in cancer as compared to normal samples in multiple cancer types. Hypervariability in these genes can be leveraged to measure deviation from a stable profile of normal expression, resulting in a cancer anti-profile.
Here we further advance this approach by demonstrating that it can also be used as a predictor of survival or relapse. We demonstrate that using genes that show consistent, or universal, hyper-variability across cancer types, their degree of deviation in gene expression from normal tissue can be used as a measurement of potential malignancy (measured as risk of relapse or death). The results indicate that the anti-profile approach can be used as a more robust and stable indicator of tumor malignancy than traditional classification approaches.
Methods
We extend the observation of consistent hypervariability in cancer with respect to the normal samples to include tumor progression. Our hypothesis here is that the degree of hyper-variability as measured with respect to the normal samples would increase with tumor progression.
Corrada Bravo et al.
12
defined how to derive a colon-cancer anti-profile for screening colon tumors by measuring deviation from normal colon samples. Briefly, to create an anti-profile, a set of normal samples and tumor samples is selected; probesets are then ranked by the quantity
We used a number of publicly available microarray datasets and two methylation datasets. The expression microarray datasets were either Affymetrix Human Genome U133 Plus 2.0 (GPL570) or Affymetrix Human Genome U133A (GPL96). The raw data were collected and processed using frozen robust multiarray analysis (fRMA) normalization
13
and the barcode algorithm
14
to obtain
For anti-profile analysis, a variance-ratio statistic across multiple tissues is calculated.
12
This statistic is computed as
The normal regions of expression are calculated based on median and median absolute deviation (mad) statistics as
The normalized expression values and the selected probesets can be supplied to the
For comparing the anti-profile scoring method against classifiers that do not take into account the hypervariability of cancer, we compared our approach with PAM, a popular shrunken centroid classifier. 16 PAM extends the regular centroid-based classification by computing a standardized centroid for each class. The shrunken centroid represents the class using the average gene expression of that class divided by the within-class standard deviation for that gene. The amount of shrinkage is determined via a threshold parameter that affects the classification by reducing the effects of features that are determined to be non-informative.
For shrunken centroid classifications, we used
Results and Discussion
Gene Expression Anti-Profiles Capture Tumor Progression
In our experiments, we first extended the anti-profile approach by using colon-cancer anti-profiles for differentiation between tumors according to their progression level. To test our hypothesis, we obtained two publicly available microarray datasets with normal, adenoma, and cancer colon samples.17,18
Based on the finding that consistent decreases in methylation are observed along large genomic blocks,
19
probesets were selected in Ref. 12 by selecting genes that lie inside such blocks to create a colon-cancer anti-profile. From those probesets, we selected the 100 probesets that showed most hyper-variability among cancer samples in comparison to the normal samples. We then plotted the distribution of variance of cancer/adenoma samples to variance of normal samples ratio (in log2 scale) for these probesets on the other dataset (Fig. 1A and B). Both adenoma and cancer samples show higher variability than normals (region to the right of
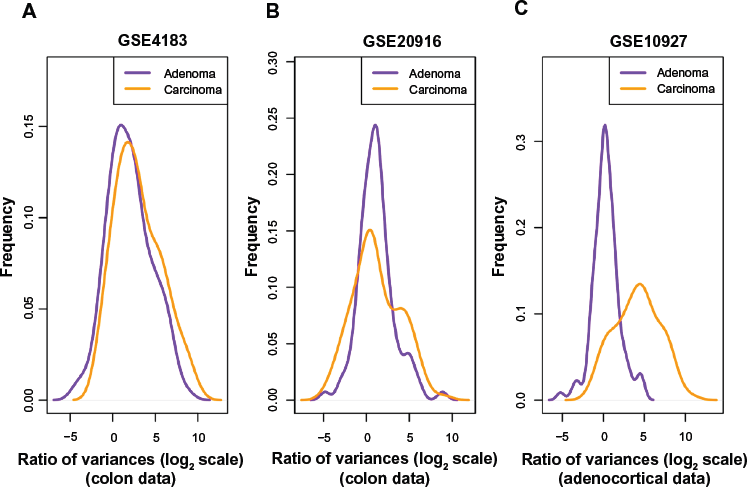
Among probes that exhibit higher variability among cancers than among normals, the degree of hypervariability observed is related to the level of progression. (
Next, we performed an analysis using the universal anti-profile signature computed in Ref.12 We obtained an adrenocortical microarray dataset
20
containing normal, adenoma, and cancer samples. For the most hypervariable 100 probesets from the universal anti-profile, we plotted the distribution of variance of cancer/adenoma samples to variance of normal samples ratio (Fig. 1C). The same observations as before could be seen: a majority of these probesets show greater variability among cancer and adenoma samples than among normal samples, and this degree of variability is higher among cancer samples in comparison with adenoma samples (A Kolmogorov–Smirnov test between the two distributions with the alternative hypothesis being that the distribution for adenoma samples is less than that of cancer samples yields
In the next stage of our experiments, we applied the anti-profile scoring method. As discussed in Ref. 12, the anti-profile scoring method counts the number of hypervariable probesets for which the expression of tumor samples lies beyond the normal region of expression. It has been shown to be an effective measurement in differentiating between tumor samples and normal samples, and our aim was to apply the same scoring method for differentiating between different stages of tumor progression. With the two colon-cancer datasets used to derive colon-cancer anti-profiles, we used the hypervariable probesets and the normal regions of expression for probesets derived from one dataset to calculate anti-profile scores for the normal, adenoma and cancer samples in the other dataset. The distribution of these scores are plotted in Figure 2A and B: for both datasets, the average anti-profile score increases from the normal group to the adenoma group to the cancer group: for the first dataset,
17
the mean scores for these groups are 18.88, 27.93, and 35.33, respectively, and for the second dataset,
18
the respective mean scores are 32.2, 51.4, and 58.9. Comparing the adenoma scores against the cancer scores yields an area under the receiver operating characteristic (ROC) curve (AUC) of 0.711 and a
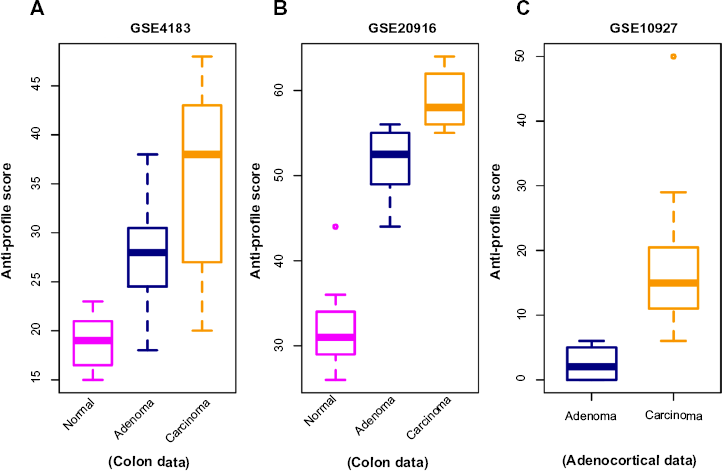
Anti-profile scores calculated for tumors and normals. (
Similarly, we applied the anti-profile scoring method for the adrenocortical dataset with the universal anti-profile probesets (Fig. 2C). The cancer samples have higher anti-profile scores than the adenoma samples: the mean anti-profile scores are 2.5 and 16.84 for the adenoma and cancer groups, respectively. The comparison of the two score groups gives an AUC value of 0.997 and a Wilcoxon rank-sum test
Anti-Profiles Based on DNA Methylation Also Capture Tumor Progression
DNA methylation is one of the primary epigenetic mechanisms for gene regulation, and is believed to play a particularly important role in cancer. 22 High levels of methylation in promoter regions are usually associated with low transcription.23,24 Abnormal methylation patterns have been observed in cancer, with loss of sharply methylation levels (in comparison with normal methylation levels) in regions associated with tissue differentiation,19,25 and is associated with increased hypervariability in gene expression across multiple solid tumor types. 25 Given these observations, we expected that the anti-profile method would be applicable to methylation measurements from samples through cancer progression.
We applied the anti-profile scoring method to DNA methylation data from thyroid and colon samples, 19 where for each tissue type, normal, adenoma and cancer samples were available. Using 384 probesets available in their custom Illumina methylation array data, for each cancer type, we used the normal samples to define the normal regions of methylation and calculated anti-profile scores by summing the number of features that fell outside the normal methylation region for each cancer sample.
Figure 3 shows the distribution of adenoma and carcinoma samples against normal samples on a principal component plot, showing the presence of the hypervariability pattern in methylation data: the normal samples cluster tightly, while the adenomas show some dispersion and the carcinomas show even greater dispersion. Since these behaviors are present for both colon and thyroid data, it again reinforces our notion that the anti-profile approach has wide application for classification in cancer.

Anti-profiles applied to methylation data: first two principal components of (
Supplementary Figure 1 shows the results obtained with the anti-profile scores. As with the gene expression data, the methylation data also show that adenomas tend to have lower anti-profile scores than the carcinomas: for the thyroid tumors, the median anti-profile score for the adenoma class is 10, while for the carcinoma class is 17, and for the colon tumors, the median score for the adenoma class is 75.5, while for the carcinoma class is 121.5.
We also obtained data from a multiple solid tumor methylation study based on the Illumina HumanMethylation450 beadarray.
25
This dataset contains DNA methylation levels for normal, adenoma, and cancer samples comprised of thyroid, breast, colon, pancreas and lung tissues (see Supplementary Table 2).
25
Sample-specific methylation levels over CpG clusters were obtained as described in the Supplementary Methods section. To test the ability of anti-profile scoring to capture stable epigenetic marks across multiple tissue types, we followed a two-stage leave-one-tissue-out cross-validation procedure for each tissue type in the dataset (see Supplementary File), where feature selection for each tissue-specific anti-profile is based on consistent hypervariable methylation within common hypomethylation blocks of the other tissues in the dataset and anti-profile construction is based only on the normal samples of the tissue. In this case, no tumor data for each tissue are used when constructing their anti-profile. We observed high separation in anti-profile score between adenoma and tumor in all tissues (Supplementary Fig. 2, Wilcoxon rank-sum test; colon,
Increased Expression Variability is Associated with Clinical Outcome in Colon, Lung, and Breast Cancer
Based on the observation that increased expression variability and anti-profile scores in probesets with hypervariable expression is associated with tumor progression, we hypothesized that it will also be associated with clinical outcomes for tumors: aggressive tumors exhibiting poor clinical outcome would be associated with increased hypervariability in these specific genes and vice versa.
Application to Colon Cancer
We first experimented with a colon-cancer anti-profile as discussed in the previous section. We obtained a microarray dataset of colon tumor samples supplied with survival information (indicating the relapse of a patient within a certain number of years). 26 Using the other colon-cancer microarray datasets used in the previous section,17,18 we constructed a colon-cancer anti-profile using the normal and cancer samples, limiting the probesets to the colon-cancer hypervariable genes from Corrada Bravo, et al. 12 A set of 100 probesets with the highest variability among cancer samples with respect to normal samples was selected from this anti-profile. These probesets and the normal regions of expression calculated for them using the normal colon samples from the abovementioned datasets together constituted the colon-cancer anti-profile used.
We stratified the samples into high risk and low risk as follows: patients who relapsed within 1 year after diagnosis were classified as high risk and those who did not relapse within 1 year were classified as low risk. For the selected probesets, we calculated the distribution of variance of high-/low-risk samples to variance of normal samples ratio (Supplementary Fig. 3). The hypervariability of the colon-cancer anti-profile probesets is reflected in these results, given that the majority of the probesets have a log2 variance ratio >0. We can also see that the high-risk samples exhibit slightly higher variability than the low-risk samples when compared against the normals, affirming that the hypervariability observation extends to tumor prognosis as well.
Further, we calculated anti-profile scores for the colon tumor samples. Since the high-risk and low-risk grouping is not a well-defined classification, only tentatively captures tumor progression, we used Kaplan–Meier survival curves to measure the effectiveness of the anti-profile scores. We ordered the tumor samples according to the anti-profile score and stratified them to three equal- sized groups and observed the rate of survival in each group using Kaplan–Meier curves. This demonstrated that the anti-profile scores clearly correlate with the prognosis of the tumors, with higher anti-profile scores showing higher chances of relapse and vice versa (Fig. 4A; log-rank test score 9.452,
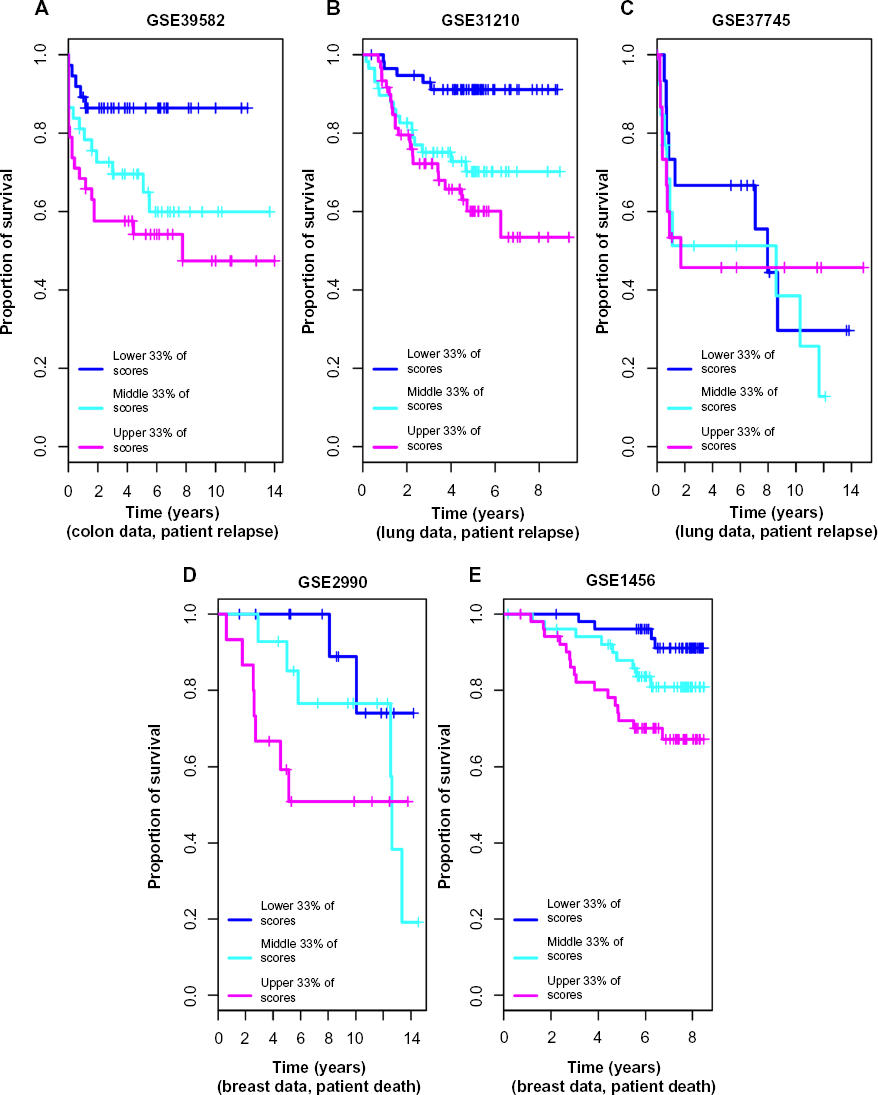
Anti-profile scores correspond to tumor prognosis: (
Application to Lung Cancer
Next, we applied the anti-profile method to analyze lung cancer survival. Here we tested the universal anti-profile from Corrada Bravo, et al. 12 with two microarray lung cancer datasets containing patient survival information based on patient relapse, the primary dataset containing both normal and tumor samples 27 and the secondary dataset containing only tumor samples. 28
As with the colon dataset, we stratified the samples into high risk and low risk based on patient relapse within five years. For the universal anti-profile probesets, we plotted the distribution of variance of high- and low-risk samples to variance of normal samples ratio (Supplementary Fig. 4). The majority of the universal anti-profile probesets show higher variability among the tumor samples than the normal samples, indicating that the universal anti-profile manages to capture the hypervariability property of these datasets.
We used normal lung samples to calculate anti-profile scores for the tumor samples for both datasets. Ordering the tumor samples by anti-profile score, for each dataset, we stratified them to three equal-sized groups and plotted Kaplan–Meier survival curves (Fig. 4B). For the first dataset, the tumor samples with the highest anti-profile scores show greatest relapse among the three groups, while the tumor samples with the lowest scores show the least relapse (log-rank statistic for the first dataset 15.44,

Anti-profiles applied to Cox proportional hazard models for survival prediction: Cox proportional hazard models with significant clinical covariates and anti-profiles were used to predict patient survival at 5 years for the second breast caner dataset (Pawitan et al) with (
The results obtained for the second lung dataset did not show as much separation in the Kaplan–Meier survival curves when sorted into three groups. Comparing the generalized normalized unscaled standard error (GNUSE) values, a standard metric of microarray quality,
29
to compare the quality of the microarray data for the two lung cancer datasets (Supplementary Fig. 6), we noticed that this second dataset has a higher GNUSE value distribution in comparison to the first dataset, which might explain the poor performance. However, stratifying the samples as top 50% of scores and lower 50% of scores did show some separation of the two groups in terms of survival (log-rank statistic 1.418,
The first dataset also contained information about death of patients. A similar analysis as before with patient death instead of relapse showed a log-rank statistic of 8.342 with
These results demonstrate that the universal anti-profile probesets can be used to model the hypervariability in lung microarray data and further validate the use of using deviation from normal samples as a measurement of tumor prognosis.
Application to Breast Cancer
We next applied the methodology to breast cancer microarray data on Affymetrix Human Genome U133A platform (GPL96). Since the universal anti-profile signature had been derived from Affymetrix Human Genome U133 Plus 2.0 (GPL570) microarray data, we used a number of publicly available GPL96 platform cancer and normal samples (1207 cancer samples and 773 normal samples) of multiple tissue types to recalculate an anti-profile signature for the GPL96 platform (see the Methods section). We used the most significant 100 probesets from this signature for our breast cancer anti-profile experiments.
After obtaining two publicly available breast cancer microarray datasets,30,31 we selected lymph node negative and estrogen receptor (ER) positive samples and verified that these probesets were able to capture the hypervariability of cancer samples (Supplementary Fig. 8). Since relapse information was not available for majority of the samples, we used death within 5 years as our criteria for obtaining a high-risk-low-risk classification.
We collected breast normal samples from publicly available datasets and calculated anti-profile scores for the two datasets. We drew Kaplan–Meier survival curves by ranking the samples by score and grouping them into three equal-sized classes (Fig. 4C). Similar to our observation with colon and lung cancer data, the anti-profile scores showed a correlation with survival of patients (log-rank statistic for the first dataset 3.971,
The second breast dataset also contained information about patient relapse. Performing a similar analysis using relapse instead of death provided a log-rank statistic of 10.755 (
In addition, Supplementary Figure 11 shows similar results obtained for the third breast cancer dataset with patient death information. With only nine deaths being recorded, our method of stratifying samples into high-risk and low-risk classes was not appropriate for this dataset. However, we observed a trend of samples with high anti-profile scores exhibiting a higher rate of relapse and vice versa, as with the other datasets.
In summary, these results obtained for lung and breast cancer data further show the utility of the anti-profile approach as a robust and effective method for modeling tumor prognosis and validate our hypothesis that deviation from the normal group can be considered a measure of the risk level associated with a tumor.
Anti-Profile Approach is More Stable than Standard Classification Methods
We compared the anti-profile method with PAM using lung cancer data. For this, using the high-risk and low-risk stratification of samples previously described, we constructed a binary classification problem between low and high risk, and trained the PAM classifier on one dataset and tested the classifier on the other dataset. We used cross-validation on the training dataset to determine the threshold parameter that minimizes the misclassification error on the training data. The same experiment was performed between the two breast cancer datasets, and also, the two colon-cancer datasets used in our analysis were based on tumor progression (here the adenoma/carcinoma status was used as the binary stratification). The posterior probabilities obtained for the testing dataset were used to calculate AUC values and Wilcoxon rank-sum test
To compare against this, we applied the anti-profile method to the same training and testing dataset pairs. For each tissue type, we used normal samples and the tumor samples of one dataset to select probesets and calculate anti-profile scores for the other dataset. A comparison of these results can be seen in Table 1.
Comparison of prediction results obtained using the anti-profile scoring method and PAM. For each tissue type of lung, breast, and colon, two datasets with tumor samples were obtained and both the anti-profile method and the PAM model were fitted on one dataset and tested on the other dataset. For a binary stratification of samples by risk level, AUC values and the p-values from the Wilcoxon rank-sum test were calculated from the decision values resulting from each method. Datasets used were Lung1(GSE31210), Lung2(GSE37745), Breast1(GSE2990), Breast2(GSE1456), Colon1(GSE4183), and Colon2(GSE15960).
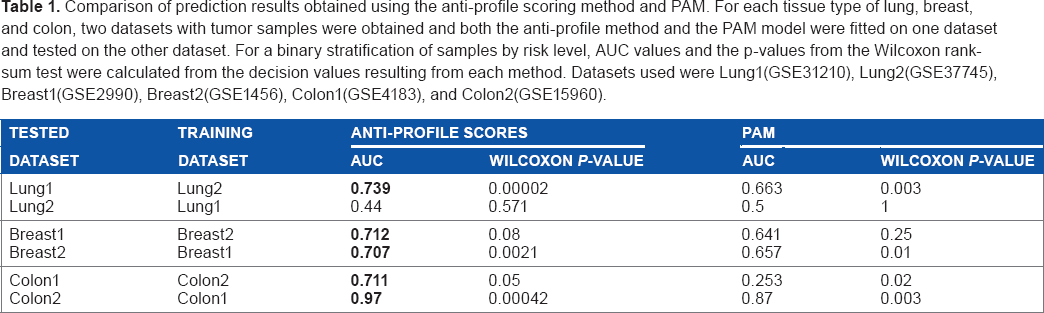
From the comparison of AUCs and the Wilcoxon rank-sum test
Anti-Profile Approach may be used for Prognostic Prediction
To further examine the prognostic ability of the anti-profile score, we used the anti-profile scores as a covariate for modeling patient survival for some of the datasets. We obtained clinical covariates for the microarray datasets when publicly available and fitted each covariate separately to a Cox proportional hazards model to ascertain their prognostic significance. The Cox proportional hazards model is a widely used statistical model for assessing censored survival information. 32 It provides a way for modeling the effect of a particular factor (such as age, severity of disease, etc) on the time taken by a patient to relapse (from the time of entering the clinical trial) or the time at which a patient dies. Here we treated the anti-profile score in the same manner as the other clinical factors.
For the first lung cancer dataset,
27
we tested age, sex, smoking status, and pathological stage. After fitting each covariate individually to a Cox proportional hazards model (assuming constant covariates) with patient relapse information, only pathological stage provided a
For the second breast cancer dataset,
31
we tested pathological stage and subtype (Basal, ERBB2, Luminal A, Luminal B, Normal Like) for prognostic relevance with relapse and found that only pathological stage was significant when fitted to a Cox model (Wald test
For the colon-cancer dataset with survival information (patient relapse),
26
we tested age, pathological stage, chemotherapy (treatment or lack of it), and location (distal vs. proximal). Pathological stage and chemotherapy status proved to be significant (Wald test
For these datasets, we also tested the predictive ability of a Cox model fitted with the covariates selected above by predicting whether a given patient would live up to a given time
For the second breast cancer dataset,
31
a Cox model fitted with the pathological stage proved to be less accurate than a model fitted with the anti-profile score (Fig. 5A) for predicting patient death at 5 years. The mean accuracy level for the model fitted with the pathological stage was 0.619, for a model fitted with the anti-profile score was 0.726, and for a model fitted with both covariates was 0.655. A Wilcoxon test between the results from the first and third models yielded
We used a similar experiment on the lung and colon-cancer datasets mentioned above, but found that adding anti-profile scores to survival models, including significant clinical covariates, did not improve their performance significantly (Supplementary Fig. 12).
Conclusions
Our aim has been to develop a robust and stable approach for classification of tumor samples. We have demonstrated that the anti-profile scoring method, which was initially applied for classification between tumor and normal samples, can be extended to classification between tumor samples as well. This method has the particular advantage that tumor samples are only used to select probesets, but given this, the anti-profile score is based strictly on normal tissue samples. The ability of the anti-profile score to successfully provide a ranking of tumor samples, which correspond to their risk of relapse (or death) and the robustness of the method across experimental datasets, demonstrates that the universal anti-profile signature provides a robust basis to develop feature selection methods for tumor prognosis and diagnosis-related microarray experiments. In addition, it confirms our hypothesis behind the extension of the anti-profile approach to tumor prognosis: the measurement of deviation from a set of normal samples, which are likely to be more cohesive, is a more stable and robust indicator of the risk level of a tumor sample as opposed to direct comparisons between the highly variable tumor samples.
High-throughput technologies for gene expression measurements, especially microarrays, have progressed to the point that the use of gene expression data to develop gene expression-based cancer signatures is quite common in cancer research. 33 However, despite a number of gene expression profile-based signatures being published and even commercially utilized, in many instances, the developed signature has performed inadequately under subsequent validations. Validations of such signatures should ideally be carried out on populations completely independent of the population selected for the derivation of the signature. Only a few gene signatures produced, such as the Amsterdam 76-gene signature, 34 have proven to be reliable for clinical use.
Heterogeneity among multiple types of tumors has been a well-known observation. 11 While the proliferating ability of tumor cells is a widely used principle behind many prognostic gene signatures, this is usually measured via a mean-shift-based differential expression measurement. However, Feinberg and Irizarry 35 demonstrate that increased variance in the genotype may increase fitness via increased variability of the phenotype, regardless of any significant change in the mean phenotype. This shifts the focus of measuring tumor heterogeneity from a mean shift to a variance shift.
As part of a comprehensive study of the colon-cancer methylome, the degree of hypervariability in DNA methylation between the adenoma and the cancer samples was observed to increase significantly. 19 When projected to a lower dimensional space using PCA, the normal samples clustered tightly together with the cancer samples dispersed and the adenoma samples demonstrated an intermediate degree of variability and an intermediate distance to the normal cluster. Based on these findings, Bravo et al. 12 introduced anti-profiles as a stable method for screening multiple types of cancer. The principle underlying this model of cancer screening is that certain genes will consistently show higher across-samples variability among cancer samples as compared with normal samples. In this study, these genes are identified and the hyper-variability is used to predict outcome, where the model is referred to as an anti-profile as it measures variation from normal behavior. The same study also demonstrated that the genes corresponding to expression hypervariability in cancer are also generally tissue-specific genes, an observation that is utilized to develop a universal anti-profile. Recent studies have looked at gene expression variability in the context of geneset and pathway discovery 36 and unsupervised construction of profiles in prostate cancer based on outlier analysis. 37
The anti-profile methods developed here are applications and extensions to the predictive setting of ideas in existing statistical methods developed to identify and model outliers in gene expression because of cancer,38,39 and other extensions are in active development. 40 These ideas are increasingly used in the analysis of epigenetic data.41,42 The general idea of using deviation from a stable class to classify between groups of anomalies is underdeveloped in the machine learning field, but should prove to be a fertile ground for the development of general methodology. 43
The results presented here confirm that an anomaly classification-based approach to gene expression and methylation-based experiments of tumor prognosis and diagnosis can be highly valuable. In summary, our work shows by application to lung cancer, breast cancer, colon-cancer and adrenocortical tumor gene expression datasets, and also to thyroid and colon methylation data, that the anti-profile approach does in fact produce models that are accurate, robust, and stable.
Author Contributions
Conceived and designed the experiments: HCB, WD. Analyzed the data: WD. Wrote the first draft of the manuscript: WD, HCB. Jointly developed the structure and arguments for the paper: HCB, WD. Both authors reviewed and approved of the final manuscript.
supplementary Materials
Supplementary Table 1.
A summary of the gene expression microarray datasets used.
Supplementary Table 2
A summary of the DNA methylation datasets used.
Supplementary Figure 1
Anti-profiles applied to methylation data: (
Supplementary Figure 2
Anti-profiles applied to Illumina Human Methylation 450 data: Distribution of anti-profile scores for adenoma and carcinoma for (
Supplementary Figure 3
Colon cancer survival analysis based on patient relapse: (
Supplementary Figure 4
Lung cancer survival analysis based on relapse: (
Supplementary Figure 5
Lung cancer prognosis is related to the anti-profile score: (
Supplementary Figure 6
GNUSE value comparison: Distribution of generalized normalized unscaled standard error values for the two lung cancer datasets, GSE31210 (Okayama et al.), and GSE37745 (Botling et al.).
Supplementary Figure 7
Additional lung cancer survival results: (
Supplementary Figure 8
Breast cancer analysis based on patient death: (
Supplementary Figure 9
Breast cancer prognosis is related to the anti-profile score: (
Supplementary Figure 10
Additional breast cancer survival results: (
Supplementary Figure 11
Additional breast cancer survival results: (
Supplementary Figure 12
Anti-profiles applied to Cox models for survival prediction: Cox proportional hazard models with significant clinical covariates and anti-profiles were used to predict patient survival at 5 years for (