
Abstract
Ovarian cancer is the fifth leading cause of death among female cancers. Front-line therapy for ovarian cancer is platinum-based chemotherapy. However, the response of patients is highly nonuniform. The TCGA database of serous ovarian carcinomas shows that ~10% of patients respond poorly to platinum-based chemotherapy, with tumors relapsing in seven months or less. Another 10% or so enjoy disease-free survival of three years or more. The objective of the present research is to identify a small number of highly predictive biomarkers that can distinguish between the two extreme responders and then extrapolate to all patients. This is achieved using the
Introduction
Background
Ovarian cancer is the fifth most deadly form of cancer for females, after lung, breast, colon, and pancreatic cancers. It is estimated that in the United States during 2015, there will be 21,290 new cases of ovarian cancer and 14,180 deaths. 1 Standard front-line therapy for ovarian cancer consists of some form of taxane (paclitaxel) coupled with some form of platinum (cisplatin or carboplatin), hereafter referred to as platinum-based chemotherapy. Patient response to front-line therapy is not uniform. Because it is not possible to monitor a patient continually to assess response to therapy, one can use progression-free survival (PFS) or overall survival (OS) as somewhat imperfect proxies for patient response. Initially, 70%–80% of patients appear to respond to front-line therapy. 2 However, based on the TCGA database of serous ovarian carcinoma, 3 ~10% of patients have PFS of seven months or less. In contrast, ~10% of patients enjoy PFS of three years or more and the rest most ultimately relapse and die of disease progression. 4
Therefore, it is imperative to be able to predict the responsiveness of ovarian cancer patients to front-line therapy. Our premise is that if there is a set of genetic biomarkers that are indicative of patient response, their influence is likely to be more pronounced at the two extreme ends of patient response. Therefore, if we succeed in developing one or more classifiers that are capable of discriminating between these two extreme cases, then these classifiers can be extended to encompass the entire patient population, which is precisely the objective of the present paper. We develop four different classifiers based on the TCGA Agilent data set and then validate them on the TCGA Affymetrix data set, as well as an independent data set from Tothill et al. 5
Current status
At the moment, CA125 is the only known biomarker to assess the effectiveness of therapy in ovarian cancer. However, CA125 levels are primarily used as a post facto measure that determines whether therapy is working and not as a
In general, most of the papers fall into one of the two categories. In the first category, the authors have a candidate biomarker in mind. The available patient pool is divided into two groups, and the mean values of the candidate biomarker across each group are computed. If there is a statistically significant difference between these mean values (using the Student's
The second approach is to apply some kind of machine learning algorithms to the data at hand, thereby obtaining a panel of biomarkers. Examples of such approaches include Ref. 11, in which 322 samples were analyzed to generate a 349-gene biomarker panel that performs very well, but when the 349 genes are reduced to 18 genes, the performance on the test data is poor, 12 in which a 300-gene Ovarian Carcinoma Index is constructed on the basis of 80 samples, which is then tested on 118 samples; and in Ref. 4, a panel of 14 genes is identified to differentiate between early relapse and late-stage relapse. It is worth pointing out that all of the abovementioned papers use some variant of the support vector machine (SVM) to find the biomarkers. Indeed, this is reasonable, as the SVM is very robust and is widely used in many application areas.
An excellent review of several studies can be found in Ref. 13. In Ref. 14, the authors started with a set of 151 DNA repair genes and identified a subset of 23 such genes that are then used to construct a score. Within the family of DNA repair genes, it has been suggested that various genes that arise in the nucleotide excision repair and base excision repair pathways, and single-nucleotide polymorphisms in these genes, have a role to play,
15
for example, ERCC and XRCC families of genes. Finally, a BRCA2 mutation is associated with improved survival and improved chemotherapy response,
16
although mutations in BRCA1 or BRCA2 are associated with enhanced risk and earlier onset of ovarian cancer.
6
A possible explanation is that responsiveness to PARP-based therapy is enhanced with BRCA mutations. A recent paper that made an extensive and thorough benchmark study concluded that no ovarian cancer gene expression signature is ready for clinical use yet.
17
In summary, there is no shortage of claimed biomarkers. However, none of these papers contains a
Contributions of the paper
In the present paper, we analyze the TCGA ovarian cancer data that consists of molecular measurements and clinical outcomes on nearly 600 serous carcinomas. We study gene expression levels as molecular measurements and PFS and OS as clinical parameters. Patients whose clinical parameters (survival, OS or PFS) are at the two extremes are identified using the user-defined thresholds, as described subsequently. Then, we apply an algorithm named “lone star” developed within our research group to extract biomarkers and an associated molecular signature that can discriminate between extreme patients with respect to these clinical parameters. Then, this molecular signature is extended to the entire patient population in the TCGA study. In this manner, we are able to develop a three-way classification procedure for assigning each patient into one of the three categories, namely, SR, medium responder (MR), and NR. We also use the discriminant function developed for the extreme responders to divide the entire patient population into two groups, namely, those with a positive score and those with a negative score. Kaplan–Meier curves are plotted for these two groups, and it is shown that the patients with positive score exhibit a clear survival advantage compared to those with a negative score.
The lone star algorithm was initially presented in Ref. 18 and is described in detail in Ref. 19. A brief description of the algorithm is given in the Approach and methods section. The source code of an MATLAB implementation of the lone star algorithm is freely available at the following URL: http://sourceforge.net/projects/lonestar/
Therefore, the algorithm can be readily used by even those unfamiliar with machine learning theory, without having to get into its inner workings. The biomarker panels developed on the TCGA data are then validated on an independent data set due to Ref. 5.
Approach and Methods
General approach
The broad approach adopted in this paper is now described. The TCGA Agilent data set consisting of molecular measurements on roughly 600 serous ovarian carcinomas
3
is chosen as the training data set, while the corresponding TCGA Affymetrix data set
3
and the Tothill data set
5
are chosen for validating the predictions. Of note, the validation data sets also consist solely of serous carcinomas. The TCGA Affymetrix data set serves to establish that our method is portable across platforms, while the Tothill data set serves to establish that our method is portable across both platforms and data sets. Given the training data set, a number
Definition of patient response
Definition of extreme responders
As stated earlier, patients whose survival is within the top
In the TCGA database, there are 565 serous carcinoma samples for which information is available on days-to-death, days-to-recurrence, and/or days-to-last follow-up. If PFS is used as the criterion, the patients with PFS ≤283 days were classified as NRs, while patients with PFS ≥574 days were classified as SRs. If OS is used as the criterion, then patients with OS ≤504 days were classified as NR, while those with OS ≥1202 days were classified as SR. Of note, in both the TCGA Agilent and TCGA Affymetrix databases, these break points produced 189 NRs, 188 MRs, and 188 SRs. Of note, the demographic features of the three classes were quite similar. This can be ascertained from the TCGA data set. However, when the classifier was applied to the Tothill data set, the labels of NR, MR, and SR were determined solely on the basis of the survival times, both OS and PFS. Consequently, the fraction of the NR, MR, and SR samples does not necessarily correspond to the 33rd percentile.
Prefiltering the feature set
There are roughly 12,000 genes for which measurements are available in all three data sets (TCGA Agilent, TCGA Affymetrix, and Tothill). While developing the classifier for the training data, it is not desirable to run the lone star algorithm using all 12,000+ genes. Some prefiltering is desirable based on the combination of two attributes: (i) the
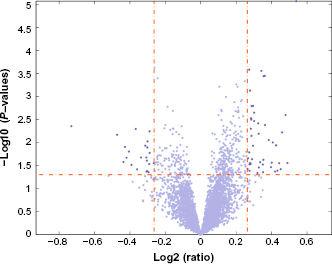
Volcano plot of the negative logarithm of the
The tight prefiltering used the following parameters: fold change of at least 1.25 between the averages of a gene's expression level over the two classes and the
The above discussion can be summarized in Table 1. There are four different combinations that are assessed in this paper: OS or PFS as clinical parameters and tight or loose prefiltering.
The four classifiers studied in this paper.

Lone star algorithm
For each of the four situations described in Table 1, the lone star algorithm was used to develop a binary classifier to identify a handful of highly predictive features, together with an associated linear discriminant function, that could be used to distinguish between the two sets of extreme responders. The following discussion is essentially reproduced from Ref. 19 in order to make the present paper self-contained.
The lone star algorithm is a very versatile and general-purpose algorithm developed in Ref. 18 and elaborated in Ref. 19, for the purpose of identifying a small number of highly predictive features from tens of thousands of measured features. It combines various ideas in machine learning, such as the
In case, we are given a set of labeled data here
In other words, the discriminant function 
Set the iteration counter to 1, the feature set
Stability selection: fix an integer


The parameter
RFE: The previous step results in
Final classifier generation: When this step is reached, the set of features is finalized. Run the
Results
Development of binary classifiers
The lone star algorithm was applied to each of the four situations, as described in Table 1. In this subsection, the details of the resulting classifiers and their performance on the
For PFS, with tight filtering and 59 genes as the starting point, the algorithm resulted in 25 genes being chosen as the most predictive features. For OS, 67 genes as the starting point resulted in 28 genes being chosen. The exercise was then repeated using a less aggressive or loose prefiltering step, so that the lone star algorithm has a larger number of initial features to choose. When OS was used as the criterion, the initial feature set consisted of 208 genes, of which 26 were finally chosen. When PFS was used as the criterion, the initial feature set consisted of 181 genes, of which 26 were finally chosen. Of note, though the number of finally selected features was comparable for all the four classifiers, the actual features themselves were different. Table 2 lists the finally selected features in the two classifiers based on OS with tight and loose prefiltering, while Table 3 lists the finally selected features in the two classifiers based on PFS with tight and loose prefiltering. In each case, the expression values of all genes are converted into
Classifier nos. 1 and 2 – classifiers for OS.

Classifier nos. 3 and 4 – classifiers for PFS.

For each classifier, receiver operating characteristic (ROC) curves were constructed by varying only the bias or threshold term to trade off between sensitivity and specificity. The resulting ROC curves are shown in Figures 2 and 3 respectively.

ROC curves with tight prefiltering. Both classifiers started with 59 initial features, of which each classifier chose 25 features (which are different from one case to the other). (A) OS as the clinical parameter and (B) PFS as the clinical parameter.

ROC curves with loose prefiltering. (A) The results using OS as the clinical parameter. The initial number of features was 208, of which 26 features were chosen finally. AUCs of the three curves are 0.8922, 0.6833, and 0.5781. (B) The results using PFS as the clinical parameter. The initial number of features was 181, of which 26 features were finally chosen. AUCs of the three curves are 0.8721, 0.6683, and 0.6886.
3 × 3 Contingency tables
The computations described in the previous subsection resulted in four different classifiers to discriminate between SRs and NRs. The next step was to use each of these discriminant functions and classify
For a 3 × 3 contingency table, the relevant quantity is the
Three-way classification based on OS and tight prefiltering.

Three-way classification based on OS and loose prefiltering.

Kaplan–Meier curves
Using the discriminant function based on the TCGA Agilent data, discriminant values were computed for all samples based on Z-scores for TCGA Agilent, TCGA Affymetrix, and Tothill data sets. This was done for all the four cases: OS with tight prefiltering, PFS with tight prefiltering, OS with loose prefiltering, and PFS with loose prefiltering. Patients were divided into two groups: with positive discriminant value and with negative discriminant value. Kaplan–Meier curves were plotted to see whether the survival (OS or PFS, as appropriate) between these two groups was statistically significant. Figures 4 through 7 show the results, including the

Kaplan–Meier curves for classifier using OS to define classes and tight prefiltering.

Kaplan–Meier curves for classifier using OS to define classes and loose prefiltering.
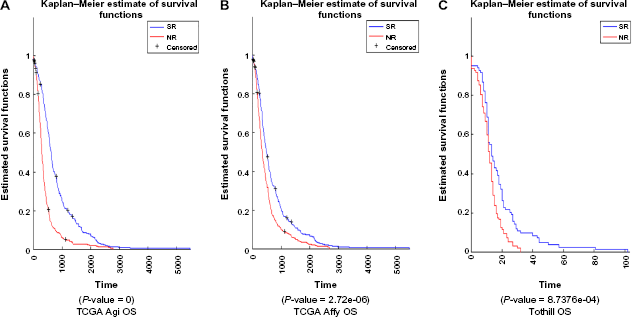
Kaplan–Meier curves for classifier using PFS to define classes and tight prefiltering.

Kaplan–Meier curves for classifier using PFS to define classes and loose prefiltering.
Discussion
We begin with a discussion of the three sets of findings, namely, the ROC curves, the 3 × 3 contingency tables, and the Kaplan–Meier curves. Then, we present an overall discussion.
From the four ROC curves, two broad conclusions can be drawn:
Classifiers based on OS to define the classes perform slightly worse than classifiers based on PFS.
The classifiers based on loose prefiltering perform better on the training data but slightly worse on the testing data.
For the 3 × 3 contingency tables, where the results of assigning
For the Kaplan–Meier curves, where the entire patient population is divided into two groups, these are the broad conclusions: on the training data consisting of the TCGA Agilent database, the group with a positive score shows a very significant survival advantage over the group with a negative score. However, on the independent validation data set, namely, the Tothill data set, once again the use of OS as the clinical parameter does not lead to satisfactory results.
In contrast, when PFS is used as the clinical parameter, the
Now we make some general comments on the outcomes of this paper. The motivation for this research was to determine whether it is possible to predict the response of ovarian cancer patients to front-line platinum chemotherapy using the biomarkers extracted in a purely data-driven fashion via machine learning algorithms. The results are mixed. From the standpoint of considerably outperforming chance, it is unmistakably clear that the biomolecular signatures based on PFS developed here perform spectacularly well on the training data set (TCGA Agilent) as well as one validation data set (TCGA Affymetrix) and also achieve
It would be highly desirable to test whether the performance on the Tothill data set could be repeated on other data sets. Unfortunately, in ovarian cancer, there very few large data sets that contain detailed information on the OS and/or PFS of patients. There is one data set, known as the Yoshihara data set, which consists of about 100 samples, and the rest contain fewer than 50 samples. With very few samples, it is not realistic to expect that classifiers would demonstrate a statistically significant improvement over pure chance. Thus, we are forced to remain content with just one independent validation data set, on which the approach leads to good results from the standpoint of statistical significance.
Along similar lines, we have not been able to locate any other molecular signature that can be readily applied to gene expression data, whose predictions can be compared with those given here. The available literature on the topic consists of biomarker panels, that is, lists of genes, but not a numerical procedure for combining the expression values of these genes to assign patients to two or more categories, as is done here.
From the standpoint of being useful in clinical practice, there is considerable scope for improvement. Ideally, the 3 × 3 contingency tables should assist the physician to assign a patient to an appropriate category. If a patient can be said to be an NR with high confidence, then she could straightaway be given alternative therapy. Similarly, if a patient can be said to be an SR with high confidence, the physician can proceed with front-line therapy in an aggressive manner. However, Tables 6 and 7 show that the positive predictive value of these categorizations
Three-way classification based on PFS and tight prefiltering.

Three-way classification based on PFS and loose prefiltering.

One of the objectives of the present paper was to compare OS with PFS as the clinical parameter to categorize a patient. It would appear a priori that OS is a more
The predictive features generated in this paper are obtained by using the lone star algorithm, 18 which does not make any use of pathway information or any other contextual information about various features. Other work carried out by a subset of the authors has led to an algorithm known as “phixer” that can be used to reverse engineer whole-genome context-sensitive gene interaction networks. Future work by our research team would consist of combining these two algorithms so as to choose features that are both highly predictive and also interpretable in terms of biological pathways.
A recent paper on melanoma 23 suggests that there are different evolutionary trajectories for different subtypes. This is a very significant observation, and it is likely that similar conclusions might apply to other forms of cancer, though this is yet to be established. If differences in patient responses in ovarian cancer were to be the result of tumors in different patients following different evolution trajectories, the complexity of the disease would increase enormously; in turn, this would make it more difficult to apply machine learning methods of the type used in the present paper.
Conclusions
In this paper, we have proposed a methodology for grouping ovarian cancer patients into three categories, referred to here as SRs, MRs, and NRs, in terms of their response to front-line platinum chemotherapy. We have also developed an approach for grouping patients into two groups in such a way that one group has a statistically significant survival advantage over the other. While both approaches achieve
Author Contributions
Conceived the problem and formulated the approach: MAW, MV. Analyzed the data and carried out the computations: BM, EA, NS. Wrote all drafts of the manuscript: BM, MV. Contributed to the writing of the manuscript: KAB, AU. Made critical revisions and approved the final version: KAB, MAW. All authors reviewed and approved of the final manuscript.