
Abstract
High-throughput transcriptome sequencing allows identification of cancer-related changes that occur at the stages of transcription, pre-messenger RNA (mRNA), and splicing. In the current study, we devised a pipeline to predict novel alternative splicing (AS) variants from high-throughput transcriptome sequencing data and applied it to large sets of tumor transcriptomes from The Cancer Genome Atlas (TCGA). We identified two novel tumor-associated splice variants of matriptase, a known cancer-associated gene, in the transcriptome data from epithelial-derived tumors but not normal tissue. Most notably, these variants were found in 69% of lung squamous cell carcinoma (LUSC) samples studied. We confirmed the expression of matriptase AS transcripts using quantitative reverse transcription PCR (qRT-PCR) in an orthogonal panel of tumor tissues and cell lines. Furthermore, flow cytometric analysis confirmed surface expression of matriptase splice variants in chinese hamster ovary (CHO) cells transiently transfected with cDNA encoding the novel transcripts. Our findings further implicate matriptase in contributing to oncogenic processes and suggest potential novel therapeutic uses for matriptase splice variants.
Introduction
Alternative splicing (AS) allows a normal cell to generate multiple pre-messenger RNA (mRNA) transcripts of a gene, which can be translated into functionally diverse proteins. Similarly, cancer cells can usurp this mechanism to tailor functional transcripts that favor the malignant state. Splice variants have been identified in a variety of cancers, suggesting that widespread aberrant and AS may be a common consequence or even a cause of cancer. 1 The biological activity of the majority of AS isoforms and, in particular, their contribution to cancer biology have yet to be elucidated. However, a number of studies have demonstrated that cancer-associated splice variants can serve as diagnostic or prognostic markers, or predict sensitivity to certain drugs.2–4 Treatments targeting these tumor-associated splice variants [eg, epidermal growth factor receptor (EGFR), CD44, and vascular endothelial growth factor (VEGF) receptor] are also showing promising results in preclinical studies and clinical trials.5,6
Massively parallel RNA sequencing (RNA-seq) allows the exploration of cancer-related changes at the level of transcription and splicing. In this study, we devised an AS-detection pipeline based on ABySS
7
and Trans-ABySS
8
software packages. ABySS is a
Matriptase (
Matriptase activity is tightly regulated via antagonism from HGF activator inhibitor-1 (HAI-1). HAI-1 is a serine peptidase inhibitor encoded by Kunitz-type 1 gene (
Matriptase is widely expressed by the epithelia of almost all organs examined so far. 22 Studies of matriptase-deficient mice have shown that matriptase is essential for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. 23 Matriptase has also been shown to be overexpressed in a variety of human cancers. In many cases, high matriptase expression levels are correlated with poor clinical outcome.24,25 In addition to matriptase overexpression, an imbalance in the ratio of matriptase to HAI-1 has been reported in late-stage tumors leading to the proposal that uninhibited matriptase activity may contribute to the development of advanced disease. 25
Although many studies present matriptase as a promising potential therapeutic target in oncology,25,26 its therapeutic use is limited by its widespread expression and essential function in normal epithelial tissues. However, a unique form of matriptase within tumor cells could potentially overcome this limitation. Using our AS-detection pipeline, we identified two novel tumor-associated spliced isoforms of matriptase in the transcriptome of primary ovarian, breast, prostate, head and neck, lung, stomach, and bladder carcinoma that were not in normal transcriptomes from the adjacent non-tumor tissue. We confirmed quantitative mRNA expression of matriptase splice variants using quantitative reverse transcription PCR (qRT-PCR) on cDNA panels obtained from an orthogonal set of tumor tissues and cell lines. Using flow cytometry, we further demonstrated the presence of matriptase splice variants on the surface of transfected chinese hamster ovary (CHO) cells with cDNA encoding these variants. Tumor association and the high frequency of matriptase splice variants within and across epithelial tumors suggest that these mutant matriptase transcripts may be of potential therapeutic value. This is the first study reporting tumor-associated transcripts of matriptase in human cancers.
Material and Methods
Obtaining Transcriptome Data from TCGA.
Raw RNA-seq data (Table 1) and clinicopathological data were downloaded from the TCGA data portal (http://cancergenome.nih.gov). Permission to access TCGA data was obtained from the Data Access Committee of the National Center for Biotechnology Information's Genotypes and Phenotypes Database (dbGAP) at the National Institute of Heath. Sample collection, library preparation, and RNA-seq were described by TCGA previously. 27 TCGA transcriptomes were generated from specimens that have not received any systemic treatment.
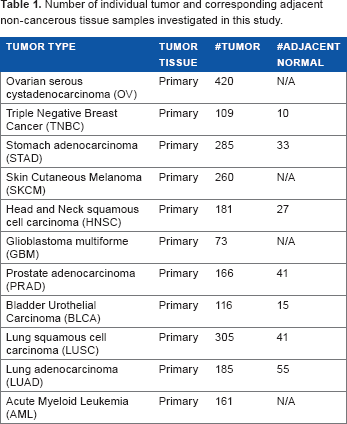
Number of individual tumor and corresponding adjacent non-cancerous tissue samples investigated in this study.
AS-Detection Pipeline.
The AS-detection pipeline starts with raw RNA-seq data (fastq files). Fastq files were either directly downloaded from the TCGA data portal or extracted from downloaded Binary Alignment/Map (BAM) files using SamToFastq.
28
The pipeline core steps include

An overview of AS-detection pipeline.
qRT-PCR Validation.
Reverse transcription reaction was performed using commercially available sets of human normal tissue or ovarian, breast, lung, and bladder cancer cDNA (OriGene Technologies); as well as cDNA synthesized from RNA isolated from ovarian cell lines, including OvCAR3, CaOV3, UACC-1598, Ov-90, and triple negative breast cell lines, including MDA-MB-231, MDA-MB-468, and HCC 1937. All cells lines were cultured under ATCC recommended culture conditions (Supplementary Method). PCR amplification was performed for 40 cycles with matriptase A1 forward 5'-GAC ACC GGC TTC TTA GCT GAA T-3’ and A1 reverse 5'-GAA GAG GGG CTT GCA GAA CTT G-3’, as well as A3 forward 5'-GAA CGA CTG CGG AGA CAA CA-3’ and A3 reverse 5-TGC TCA AGC AGA GCC CAT T-3’ primers. For each target, relative levels of expression were normalized against housekeeping gene beta-glucuronidase signal (GusB), generating δCt value for each reaction (Supplementary Method). GusB was found to be stably expressed across ovarian samples for use as a reference gene by Li et al. 29 The relative fold change for each sample was calculated similar to the approach taken by Beillard et al. 30 Samples were grouped by cell lines, cancer subtypes, or normal tissues, and graphed using GraphPad Prism software version 5.0 (GraphPad Software Inc.).
Transfection Constructs.
Total RNA was isolated with the RNeasy Mini Kit (Qiagen) from MDA-MB-468 and HCC 1937 cells to generate cDNA encoding HAI-1 and wild-type matriptase, respectively. cDNA was generated as per manufacturer's instructions using SuperScript® III Reverse Transcriptase (Life Technologies) and Oligo(dT)18 primer (Thermo Fisher Scientific). HAI-1 and wild-type matriptase were amplified from the above cDNA using Q5® Hot Start High-Fidelity DNA Polymerase (New England Biolabs).
Cell Culture Conditions and Transfection.
CHO-K1 cells (ATCC) were maintained in Ham's F-12 media (Life Technologies) supplemented with 10% fetal bovine serum (FBS; Life Technologies) at 37 °C and 5% CO2. The day before transfection, 2.5 × 10
6
cells per plate were seeded in the above media on four 10-cm plates for each transfection. The four transfections consisted of empty pTT5 vector alone, wild type plus HAI-1, variant A1 plus HAI-1, and variant A3 plus HAI-1. Twenty-four hours later, each transfection was performed by mixing a total of 10 μg of cDNA into 500 μ
Flow Cytometry.
Twenty-four hours after transfection, the plates were washed once with phosphate buffered saline (PBS, Life Technologies), and the cells were dissociated from the plate with non-enzymatic cell dissociation solution (Sigma-Aldrich). After 15 minutes at 37 °C, the cells were collected by pipetting up and down in PBS plus 1% FBS (PBS/FBS), counted on a Vi-Cell™, and resuspended in PBS/FBS. Cells were added to a 96-well plate, spun at 400 ×
Immunoprecipitation and Western Blot Analysis.
The immunoprecipitation was performed as described
31
with the following modifications. Unless otherwise stated, all reagents were purchased from Sigma. As outlined above for the flow cytometry experiment, HAI-1 plus either wild type, A1, or A3 transfected CHO-K1 cells were dissociated from 10-cm plates with non-enzymatic dissociation solution, and collected by pipetting up and down in PBS alone. Cells were spun for five minutes at 400 × g, and the supernatant was aspirated. Pellets were resuspended in 0.5–1 mL of ice-cold lysis buffer [50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 1 mM CaCl2, 1 mM MgCl2, and one complete mini ethylenediaminetetra-acetic acid (EDTA)-free protease inhibitor cocktail tablet (Roche) per 10 mL of buffer]. While on ice, the cells were broken open with 10 strokes of the pestle using a pestle and microtube set (VWR), and then the lysate was passed through a 26-gage syringe 10 times to shear the DNA. DNase was added to 10 μg/mL, and the lysates were gently rotated at 4 °C for 30 minutes. Lysates were clarified by centrifugation at 20,000 ×
Statistical and Survival Analysis.
For each tumor set, Kaplan-Meier survival curve of patients was prepared according to the presence status of matriptase A1 and A3 transcripts, with differences in overall survival rates determined by the log-rank test. Overall survival time was defined as the period between initial pathologic diagnosis and the time of death. Survival time of patients who were still alive was noted with the data of the most recent follow-up appointment. The Fisher's exact test was used to compare the two categorical variables. The Mann–Whitney
Results
Epithelial-Derived Tumors Express Novel Splicing Variants of Matriptase.

Schematic presentation of novel matriptase AS transcripts. Four LDL receptor class A domains are found in matriptase (LDLRA1: residues 452–486, LDLRA2: residues 487–523, LDLRA3: residues 524–561, and LDLRA4: residues 566–604). A1 and A3 are produced by skipping exon 12 (encoding LDLRA1) and exon 14 (encoding LDLRA3), resulting in in-frame deletion of 105 and 114 bp, respectively. CAT: serine protease catalytic domain.
An estimation of A1 and A3 transcript abundances using the number of reads supporting the novel exon–exon junction from Trans-ABySS indicated higher expression for A1 compared to the A3 transcript in all tumors studied (Supplementary Figs. S1 and S2). We observed a wide range in the frequency of epithelial tumors displaying these matriptase splice variants, from 3% in prostate adenocarcinoma (PRAD) to 69% in lung squamous cell carcinoma (LUSC) (Fig. 3). Matriptase variant A1 was found more frequent than A3 across all tumors studied (

Frequency of novel matriptase AS transcripts across tumors studied. Samples expressing matriptase novel transcripts were divided into three groups: (1) expressing transcript A1, (2) expressing transcript A3, and (3) expressing both A1 and A3 transcripts. Matriptase transcript A1 is more frequent than A3 (
The human matriptase is located on chromosome 11q24–25, spanning a genomic region of 50 kb. It is composed of 19 exons (NCBI reference sequence GenBank: NM_021978), and encodes a protein containing 855 amino acids. Our nucleotide sequence analysis revealed that A1 was produced as a result of skipping exon 12. Similarly, the A3 deletion occurred by skipping exon 14 (Fig. 2). Analysis of predicted protein sequences revealed that both matriptase variants contain fully functional ORFs, suggesting the possibility of expressing two novel proteins (Supplementary Sequence S2). Protein domain prediction further demonstrated that matriptase variants A1 and A3 lack LDLRA1 and LDLRA3 domains, respectively. Pairwise protein sequence alignment versus wild-type matriptase showed that the predicted protein for A1 transcript skips amino acids 452–487 followed by occurrence of an amino acid arginine (R) through the resultant of a novel exon–exon junction (Supplementary Sequence S3.1). The protein product of A1 transcript contains 820 amino acids. The A3 transcript encodes a protein of 817 amino acids, which is the result of skipping amino acids 524–562 followed by substitution of methionine (M) as a result of the formation of a novel exon–exon junction (Supplementary Sequence S3.2).
Matriptase Splice Variants are Novel and Tumor Associated.
To search AS information for matriptase, we performed literature searches using PubMed, online mendelian inheritance in man (OMIM), and other databases of AS, including the AS and Transcript Discovery (ASTD) database. 34 In addition, we searched publicly available expressed sequence tag (EST) and mRNA databases including GenBank, Ensembl, dbEST, and UniGene. Our search did not find these novel matriptase variants. We only found three AS transcripts of matriptase, which are formed as a result of an intron retention event (Ensembl IDs: ENST00000530532, ENST00000524718, and ENST00000530376). Furthermore, we did not detect the novel transcripts of matriptase in adjacent non-cancerous tissue from TCGA or in the transcriptome data available from the BodyMap 2.0 project, thus suggesting these variants are tumor associated.
qRT-PCR Analysis Confirms Differential Expression of Novel Matriptase Transcripts in Epithelial-Derived Tumors.
To validate the expression of matriptase splice variants in epithelial tumors, we designed matriptase wild type or splice variant-specific probes for qRT-PCR (Material and methods). qRT-PCR was carried out on orthogonal panels of cell lines, and human primary and metastatic tumor tissues from ovarian, breast, lung, and bladder cancer and a panel of normal tissues. The normal panel included 48 healthy tissues (Supplementary Table S2) from normal ovary, lung, bladder, and breast. We measured changes in the gene expression by comparing the threshold cycle (Ct) of PCR product detection normalized against a reference gene transcript. The expression levels detected by qRT-PCR for wild-type matriptase and its splice variants showed that wild-type matriptase was the predominant transcript in both tumor and normal tissues (
We further tested the expression of matriptase splice variants in a panel of normal tissue samples including 48 normal tissues from across the human body. Both matriptase splice variants A1 and A3 showed higher expression in tumor samples compared to the normal tissue panel (
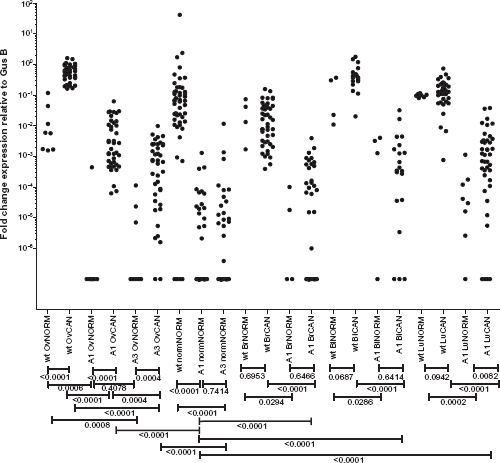
qRT-PCR validation. qRT-PCR was carried out on orthogonal panels of cell lines and human primary and metastatic tumor tissues from ovarian, breast, lung, and bladder cancer and a panel of normal tissues. Mann–Whitney
Matriptase Splice Variants Can be Translocated to the Surface of Transfected CHO Cells.
To address the question of whether matriptase A1 and A3 transcripts yield protein variants that are capable of being translocated to the cell surface, we transiently transfected CHO cells with cDNA encoding these genes followed by flow cytometric analysis of surface matriptase proteins (wild type, variant A1, and variant A3). For this experiment, we used a human anti-matriptase antibody that binds to the catalytic domain of all three matriptase variants and is not variant specific. Co-expression of the matriptase variants with HAI-1 resulted in a significant increase in the mean fluorescent intensity for wild type, variant A1, and variant A3 (

Flow cytometric analysis reveals surface expression of matriptase splice variants. Cells were transfected with 10 μg of empty vector alone (pTT5) or 5 μg of each matriptase variant plus 5 μg of HAI-1 (
Discussion
AS is a widespread mechanism for the generation of diverse protein products and regulation of protein expression. Tumor cells exploit this mechanism to favor the malignant state.1,35,36 In the past decade, cancer-associated splice variants of genes that control mechanisms such as DNA damage and proliferation [EGFR, fibroblast growth factor receptor 3 (FGFR3), breast cancer 1 (BRCA1)], adhesion and invasion [CD44, macrophage stimulating 1 receptor (MST1R)], angiogenesis (VEGF), and apoptosis [B-cell lymphoma/leukemia 10 (BCL10), caspase 2 (CASP2)] have been reported. 37 Among these, AS transcripts with altered protein structure localized to the cell surface are of particular interest as they represent potential biomarkers for discrimination between healthy and cancerous cells. That is, monoclonal antibodies can be produced to selectively target cancerous cells expressing such protein isoforms. An antibody against a tumor-associated surface-localized variant of EGFR (EGFRvIII) with exons 2–7 deleted has shown effective anti-tumor activity in preclinical studies, 6 and is now in phase I clinical trials.
With the advent of massively parallel RNA-seq, the large-scale exploration of cancer-related changes at the stage of transcription and posttranscriptional splicing has the potential to determine many more tumor-associated or enriched targets. In the current study, we devised an AS-detection pipeline from high-throughput RNA-seq data. The AS-detection pipeline allowed us to mine large sets of tumor transcriptomes to identify novel tumor-associated AS variants. Most notably, we identified two novel tumor-associated splicing variants of matriptase through analysis of more than 2,200 tumor transcriptome data available from TCGA. The variant designated A1 has an in-frame skipping of exon 12, and variant A3 is generated as a result of skipping exon 14. Our analysis revealed a high frequency of these variants across epithelial-derived tumors, which were absent or expressed at extremely low levels in transcriptomes derived from normal tissues. Novel matriptase isoforms appear to form 2–8% of the overall matriptase gene expression in tumor samples, with wild type being the dominantly expressed form (Supplementary Figs. S1, S2, and S12). qRT-PCR confirmed mRNA expression of matriptase variants, and revealed differential higher expression of variant A1 in ovarian and lung tumor tissues and cell lines compared to low or no expression in normal samples. Similarly, the A3 transcript was overexpressed in ovarian tumor tissues and cells. We then investigated variants A1 and A3 in cDNA panels derived from 48 healthy tissue types from across the body, such as brain, heart, kidney, and lung. We observed no mRNA expression of matriptase variants in more than two-thirds of normal samples and a low level of expression in the remainder. Sequence analysis indicates that the transcript variants can produce two fully functional ORFs. Our immunoprecipitation results show that these two novel proteins are being produced in CHO cells transiently transfected with cDNA encoding matriptase splice variants. With matriptase localized to the cell surface, there is a possibility that these novel isoforms of matriptase are also present on the cell surface. We tested this hypothesis by performing flow cytometry on CHO cells expressing these recombinant proteins. This analysis demonstrated the presence of these novel proteins on the surface of CHO cells, where wild-type matriptase surface expression predominated followed by variant A1 and then variant A3. Thus, protein expression of matriptase splice variants on the surface of CHO cells supports the notion that A1 and A3 protein products can localize on the surface of tumor cells as well.
The LDL receptor class A domain is an ∼40-amino-acid-long structure. The prototype structure of the LDLRA domain is found in the LDL receptor itself, which contains seven such domains. The crystal structure of the fifth LDLRA domain in the LDL receptor revealed that this domain contains six amino acids that bind calcium in an octahedral arrangement (calcium cage). 38 Point mutations at critical residues in this calcium cage have been found to potently inhibit the LDLRA ligand binding. 39 Oberst et al showed that mutations in the Ca2+-binding motifs of any or all of the four LDLRA domains of matriptase prevent its activation. 20 Interestingly, however, the complete deletion of all four LDLRA domains allows constitutive activation of this enzyme. Additional experiments are required to demonstrate the impact of deleting LDLRA1 and LDLRA3 domains as observed in the A1 and A3 variants. Although these two deletions may have variable effects on matriptase activity, our results demonstrate that they do not affect the ability of their protein products to form a complex with HAI-1 and traffic to the cell surface.
We identified no splice-site mutation associated with skipping exons 12 and 14 of matriptase in TCGA mutation analysis data derived from matching whole-exome sequencing dataset. We predicted RNA-binding proteins (RBPs) that possibly bind to matriptase mRNA using RBPmap online web server
40
(Supplementary Table S7) and compared the expression of these RBPs according to the expression status of matriptase variants. This analysis revealed significant change (
In the current study, we devised an AS-detection pipeline and performed our discovery analysis on a large number of tumors from TCGA. Our analysis revealed two novel tumor-associated splice variants of matriptase, which were confirmed in an orthogonal set of tumor tissues and cell lines. This approach highlights high frequency of matriptase variants among patients with epithelial-derived tumors as well as low or no occurrence in normal tissue. In addition to gene expression data, our flow cytometric analysis confirmed protein expression of both matriptase variants on the surface of CHO cells, suggesting matriptase variants as potential biomarkers of tumor cells. Clinical validation would prove valuable in confirming the utility of matriptase variants for therapeutic use.
Author Contributions
Conceived and designed the experiments: DD, JSB, SJMJ. Analyzed the data: DD, RDS, LY, PJB, BJH, EMD, AHM, RD, JA. Wrote the first draft of the manuscript: DD. Contributed to the writing of the manuscript: DD, RDS, LY, PJB, BJH, AHM. Agreed with manuscript results and conclusions: DD, RDS, LY, PJB, BJH, EMD, AHM, RD, JA, JSB, SJMJ. Jointly developed the structure and arguments for the paper: DD, RDS, SJMJ. Made critical revisions and approved the final version: DD, RDS, LY, PJB, BJH, EMD, AHM, RD, JA, JSB, SJMJ. All authors reviewed and approved the final manuscript.
Footnotes
Acknowledgments
The results published here are in whole or part based upon data generated by TCGA pilot project established by national cancer institute (NCI) and national human genome research institute (NHGRI). The authors would like to thank TCGA group for making these data publically available. Information about TCGA can be found at ![]() . They would also like to thank Mitacs-Accelerate for a PhD fellowship to DD. This work was carried out in facilities supported by the Centre for Drug Research and Development (CDRD), CDRD Ventures Inc. (CVI), and Genome Sciences Centre.
. They would also like to thank Mitacs-Accelerate for a PhD fellowship to DD. This work was carried out in facilities supported by the Centre for Drug Research and Development (CDRD), CDRD Ventures Inc. (CVI), and Genome Sciences Centre.