
Abstract
Various studies have implicated different multidomain proteins in cancer. However, there has been little or no detailed study on the role of circular multidomain proteins in the general problem of cancer or on specific cancer types. This work represents an initial attempt at investigating the potential for predicting linkages between known cancer-associated proteins with uncharacterized or hypothetical multidomain proteins, based primarily on circular permutation (CP) relationships. First, we propose an efficient algorithm for rapid identification of both exact and approximate CPs in multidomain proteins. Using the circular relations identified, we construct networks between multidomain proteins, based on which we perform functional annotation of multidomain proteins. We then extend the method to construct subnetworks for selected cancer subtypes, and performed prediction of potential linkages between uncharacterized multidomain proteins and the selected cancer types. We include practical results showing the performance of the proposed methods.
Introduction
Given the complex nature of multidomain proteins, it comes as no surprise that they will be involved in very complicated diseases such as cancer. Various studies have implicated different multidomain proteins in cancer. Examples here include the BRICHOS superfamily,1,2 and the BCL-2 family.3–5 However, to our knowledge, there has been little or no detailed study on the role of circular permutations (CPs) in multi-domain proteins in cancer. Yet, circular proteins and CPs in proteins are becoming of increasing interest, especially given their role in the structure, function, folding, and stability of proteins.6,7 In a circular (or cyclic) protein, the traditional N- and C-termini are joined, resulting in a protein sequence with neither a beginning nor an end. The cyclotides is a typical example of a naturally occurring family of cyclic proteins in the plant kingdom. Cyclotides are known to play a major role and provide important functions in terms of plant defense against insects and other pathogens.7–9 Their cyclic structure is known to be an important factor in their unusual stability. 7 Other common examples of cyclic proteins are the bacteriocins, small antimicrobial peptides with 30–70 residues produced by bacteria,10–12 cyclosporins found in fungi, 13 and the primate rhesus θ-defensin-1 14 with antibacterial properties for the immune system of macaque monkeys.
A CP involves the modification of a protein, first by joining the N- and C-termini to form a circular protein, and then creating a new N- and C-termini by splitting the circular protein at a different location. Thus, the new sequence formed will be a circularly permuted version of the original sequence. The earliest observation of naturally occurring CPs in proteins was reported by Cunningham et al,15,16 who showed that the amino acids of the protein concanavalin A (con A) was a CP of another homologous protein, lectin favin. Lindqvist and Schneider 17 listed several other example proteins with CPs, such as bacterial β-glucanases, α-1,3 and α-1,6 glucansynthesizing glucosyltransferases, transaldolose, the C2 domain, and saposins with a structure similar to the bacteriocins.10,11 CPs in DNA methyltransferases were earlier studied by Jeltsch and Bujnicki.18,19 Since then, various other CPs have been found in a diverse family of proteins, involved in a diverse array of functions.
Block rearrangements based on domains are common in protein evolution and adaptation.6,20,21 Thus, CPs can also occur at the block level, in terms of protein domains, rather than just protein sequences. Weiner et al. 22 argued that the conservation of catalytic centers and structural elements in artificial permutations that maintain the same function as the original sequence suggests that CPs are more likely to be block-based at the level of functional domains, rather than at the level of amino acid sequences. Thus, they proposed an algorithm for detecting domain-level CPs in multidomain proteins.22,23 Han et al. 24 reported that multidomain proteins occupy >50% of all proteomes, with eukaryote proteomes containing a higher proportion of multidomain proteins than prokaryote proteomes. The preponderance of multidomain proteins in complete genomes,25–27 and the rate at which complete genomes of several organisms are being sequenced, provides another important motivation for a deeper study of CPs in multidomain proteins.
Figure 1 shows examples of multidomain zinc finger protein sequences that are related by CPs, along with their domain block structures. In this figure, one protein (ZNF146) appears as an exact CP inside the other (ZNF680). Also, both proteins form a pair of matching 1-approximate CP. We note that, without considering CPs, these matches cannot be found using standard exact or approximate pattern matching.

Example of multidomain proteins that are related by CP. Multidomain protein Q5RFP4 (Zinc finger (ZNF146) from
There is still a debate on the origins, evolution, and prevalence of naturally occurring CPs in proteins. Various mechanisms have been suggested 22 based on evolutionary genetic events, such as duplication and deletion, 18 fusion/fission events, 22 and “cut-and-paste” mechanism 19 involving plasmids. Others have proposed post-translational modifications. 16 Craik 7 described other possible mechanisms. Further, the complete role of circularization in proteins is not yet fully understood. 7 However, circular proteins have been known to be involved in several important functions, such as plant defense against insects and other pathogens,7–9 providing stability, 7 and support of antibacterial activities for the immune system in macaques monkeys. 14 Cyclization was suggested to be critical for certain activities of the cyclic proteins, as engineered acyclic permutants of naturally occurring proteins with the same general structure were shown to exhibit loss of hemolytic activity. 29 The C2 domains (which are topologically distinct from Synaptogamin I but related by CPs) 30 are known to be involved in signaling and transduction in eukaryotes, 17 and thus could play a role in certain cancers. The WD-Repeat protein (WIPI protein family) is implicated in various human cancers, such as skin, kidney, and pancreatic cancers. The WIPI family contains beta-propellers with ring structures, which are stabilized by CPs. 31 The PDZ domain is another multidomain family that is involved in cancer. 32 Folding and misfolding of CP variants of the PDZ domain and the impact on the stability of their structure and function were studied by Hultqvist et al and Ivarsson et al.33,34 Chemically synthesized retrocyclin, a defensin-like molecule, was found to possess possible anti-HIV properties.8,35
Given the growing importance of cyclization and CPs in proteins, there is a need for efficient algorithms for their detection and analysis. Further, the preponderance of multidomain proteins, coupled with the prevalence of CPs in such proteins underline the importance of considering multidomain proteins in such an algorithmic study. For block-based multidomain proteins for instance, there are key challenges posed by the specific nature of the domain sequences, such as the very large alphabets involved, and the variability in sequence lengths (in ProDom, the multidomain protein database, 28 sequence lengths vary from as small as 2 domains, to as large as 568, with an alphabet size of almost 2 million). Most of the available algorithms for detecting CPs are still relatively slow, often running in times that are quadratic or cubic with respect to the total length of the sequences in the database. With such algorithms, an all-against-all search of possible CPs of a protein contained within other proteins becomes almost infeasible, even with multiple processors. The exponential growth in the size of available genomic datasets and the rapidly increasing rate at which complete genomes are being sequenced imply an urgent need for improved algorithms for whole-genome analysis of cyclic permutations in proteins. Such algorithms should be robust and efficient on both the direct protein sequences and on block-based multidomain representations with vastly increased alphabet sizes. They should be able to support sophisticated searches and comparisons, such as the all-against-all CP problem.
In this paper, we first propose algorithms for rapid detection of CPs in multidomain proteins, suitable for scanning large genomic databases for all-against-all circular pattern matches. Building on the results, we study networks of multi-domain proteins constructed based on their shared CPs. Using this network, we investigate a method for functional annotation of uncharacterized multidomain proteins. We then extend the method to study potential association of some unknown multidomain proteins with certain types of cancer.
Background and Related Work
Basic notations
Let
Circular pattern matching
Computing similarity (or dissimilarity) between two strings is an important problem in general sequence analysis,36–38 pattern recognition,39,40 and biology.41,42 The major computational tool used to study CPs is based on solutions to the circular pattern matching (CPM) problem.
Two strings are CPs of each other if one can be transformed to the other through a sequence of circular shifts. A circular shift is a mapping
Algorithms for the CPM problem have been proposed for the exact CPM problem,36,43,44 and for the
CPM problems in protein sequences
A number of studies have been reported on algorithms for detecting CPs for protein sequences.41,42,50 The first method
51
used the dot matrix and human visualization to identify circular relationships between pairs of protein sequences. Altschul et al
52
used a dictionary method to find short fragments common to the protein sequence pairs and used human visualization to report the best local matches. Uliel et al.53,54 introduced a method to detect CPs in protein sequences using global alignment.
55
They gave an
More fundamentally, both groups22,23,53,54 that have studied CPM in protein sequences have focused on whole sequence comparison with another whole sequence. In their experiments, they have to group the protein sequences based on their specified lengths, and used the dissimilarity in lengths for initial pruning. These methods ignored the fact that a shorter circular protein sequence could be part of the functional region of a much larger multidomain protein. This, however, could be a key consideration in function prediction for multidomain proteins. Further, as with the more theoretical algorithms for the ACPM problem,45–47 the methods for protein sequences22,23,53,54 also only considered the existential version of the ACPM problem (ie, simply report
Other recent work on CPs have studied structure alignments for circular proteins.57–61 Our focus is on rapid and efficient search for CPs, rather than on alignments. We address the enumerative version of the ACPM problem, and use the results to study functional associations between multidomain proteins. We also apply our results to the problem of predicting cancer-related multidomain proteins. Our circular pattern detection method is based on a very different approach, using indexing on suffix arrays.
Materials and Methods
Datasets
The major sources of data used are the protein domains in the ProDom database, protein annotation in the gene ontology (GO) database, and information on proteins with known association with cancer.
Protein domain database
Most proteins consist of several domains. The same protein domain may occur in many related proteins. Our experiments were performed using multidomain proteins in ProDom, 28 a database of known protein domains. Each domain is represented as a unique symbol, thus a multi-domain protein is viewed as a sequence of such symbols. The length of the domain representation is generally much smaller than the original protein sequence, but the size of alphabets has increased drastically. Pagel et al. 62 constructed a protein domain interaction network using data from ProDom.
GO database
The GO project (http://www.geneontology.org/) provides a description and annotation of genes and protein products in different databases including the known functions of the genes. Currently, the GO Consortium includes many databases such as GeneDB (http://www.genedb.org/), UniProtKB-Gene Ontology Annotation (UniProtKB-GOA) (http://www.ebi.ac.uk/GOA/), and FlayDB (http://flybase.bio.indiana.edu/). The ProDom database provides the Accession Number for the parent protein of each domain. The Accession Number is also provided for UniProtKB-GOA. This establishes a connection between entities in ProDom and their corresponding entities in GO. Thus, we can use this relationship to obtain the GO terms used to describe the protein function.
Cancer Protein Datasets
The Cancer Resource
63
is a database of proteins known to be associated with cancer. The database contains information on 25 general cancer categories. For our experiments, we downloaded and analyzed protein data on five cancer categories, namely, bone, colon, lung, skin, and breast. The Cancer Resource dataset is available at on the web (http://bioinf-data.charite.de/cancerresource/). To verify some of the novel cancer-related proteins predicted by our algorithm, we performed literature search using PubMed, and also checked for entries in the publicly available
Algorithms for CPM
In this section, we present our algorithms for the ACPM problem. First, we introduce APM-via-LIS (
The LIS method utilizes the LIS algorithm36,64 to calculate the longest common subsequence (LCS)36,65 between two sequences. The verification process checks whether the edit distance between these two sequences is less than
For each matched symbol, we obtain its positions of occurrence in the two sequences. We can use these positions to check the number of edit operations between two matched symbols. Thus, the algorithm reports the edit distance between these two sequences. The time complexity for this algorithm is

Generic approximate pattern matching using LIS APM-via-LIS(
Algorithm 1: Greedy ACPM Algorithm
Algorithm ACPM-greedy (
This algorithm is simple, but greedy (suboptimal): it finds only one occurrence of the pattern, and may not detect all the circular patterns that occur in the text. If there is more than one LCS in

ACPM with Greedy Algorithm ACPM-GREEDY(
Time complexity analysis
For the complexity analysis, we need to consider two cases: (1) For the case of searching for one sequence against a group of sequences (loop from line 2 to line 6), the time complexity is
Algorithm 2: ACPM with q-grams and suffix array
The
Figure 2 shows the number of hypotheses generated for different
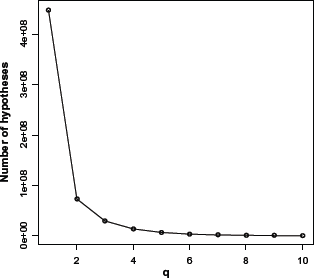
Variation of the number of hypotheses generated using
Algorithm ACPM-QGRAM (Algorithm 2) shows the process. Lines 1–7 denote the preprocessing stage. This stage constructs a long concatenated sequence,
Lines 9–24 use a loop to generate all the hypotheses for the
Lines 15–22 perform the verification. We use the ACPM-via-LIS algorithm to verify the approximate patterns. Constructing the circular pattern is the same as in the previous algorithm. We enumerate the

ACPM with q-grams and Suffix Array ACPM-qgram(
Complexity analysis
The required suffix array for the entire database can be constructed in
Comparison with other ACPM algorithms
The time complexity of our ACPM-QGRAM algorithm is
Multidomain protein networks using circular patterns
We explore the use of our proposed CPM algorithms on the problem of analyzing multidomain protein sequences. Based on the circular patterns found by our algorithms, we construct a multidomain protein network by connecting different multidomain proteins that are found to be associated by some matching circular patterns. We note that Pagel et al. 62 introduced a tool for analyzing potential relationships between proteins using the protein domain network. This network was based on protein domain interaction networks. They built a web resource to explore Domain Interaction MAp (DIMA). In this network, the nodes are the protein domains and the edges are the interactions between two protein domains. In our work, network formation is based primarily on cyclic relationships between multidomain proteins.
More specifically, we construct a directed graph showing a relationship network among the multidomain proteins. Each node (vertex) in the network represents a multidomain protein, while an edge between two nodes represents a circular relationship between the nodes. For a given node, we define the
Protein function prediction
The network described above provides an important framework for studying potential functional linkages between the multidomain proteins in our dataset. First, Table 1 provides an intuition on how an analysis of the network of CP relations could expose potential associations between multidomain proteins. The table shows the protein functions for the Top 20 highest degree proteins. We notice that 15 of the 20 proteins have exactly the same functions. Four of the proteins (ranked 3, 4, 9, and 12, respectively) do not have entries in the GO database. Protein Q5NU40 (rank 14) has a record in GO database, but no function has been assigned to it in GO. Thus, the functions of these five proteins are not yet known. It is expected that some of these five proteins with no known function are likely to have the same or similar functions as the other 15 proteins.
Top 20 highest degree proteins with GO function.

We use the
Experiments and Results
Setup
We performed experiments using the results of the proposed algorithms to study CPs in multidomain proteins, looking for potential CP relationships between pairs of such proteins. We use the results of the proposed algorithms to study potential functions linkages between uncharacterized or unknown proteins and known multidomain proteins. In the experiments, each domain is a symbol in the alphabet. Thus, |Σ| is quite large, and often in
The experiments were performed using a DELL PC, with 4 x 2.67 GHz CPU, and 8G memory, running Ubuntu 10.10 Linux operating system. All programs were compiled using gcc.
Speed and completeness
We ran the three proposed algorithms on the ProDom dataset and use the results to analyze the relationship between multidomain proteins. The ACPM-QGRAM algorithm was executed using two different parameter settings, namely
ACPM-GREEDY algorithm is faster than the other ACPM algorithms, but the result has low accuracy (around 50%). Algorithm ACPM-LIS was the slowest algorithm. Figure 3 shows the practical time required by these three algorithms, where the
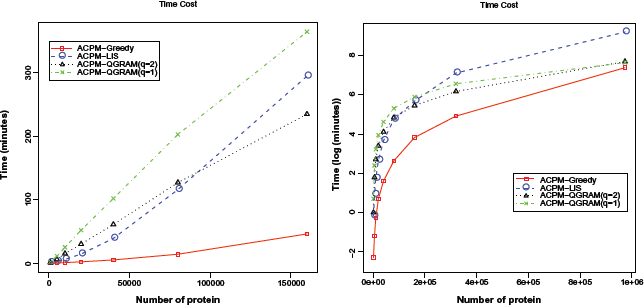
Execution time for the proposed ACPM algorithms.
We also implemented a hybrid algorithm where an exact CPM (ECPM) algorithm
48
was applied first and then followed by the ACPM-QGRAM algorithm with parameter
Circular patterns found in the ProDom database.

Statistics of CP Network in ProDom
First, we investigate the nature of the multidomain protein network formed between protein that share some CPs in the ProDom dataset. Figure 4 shows the log plot of the degree distribution for the network. Figure 4A shows the degree distribution for all vertices in the network, while Figure 4B shows the degree distribution of the Top 100 highest degree nodes.
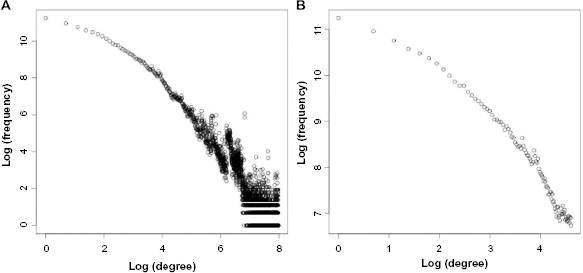
Degree distributions in the network of multidomain proteins constructed based on the circular patterns they contain. (A) Log degree distribution. (B) Log degree distribution for Top-100 degree nodes.
The results show the power and significance of our basic approach, addressing the all-against-all variant of the ACPM problem. Each protein sequence is not only used as a pattern to search against the other protein sequences, but also used as text to be searched on using the other protein sequences in the database. Of the 973,686 multidomain proteins remaining in our dataset after preprocessing, 799,044 (85%) contain at least one other protein sequence as a circular pattern; 424,888 protein sequences (43.6%) were found to be a pattern in some other protein sequences; 374,279 protein sequences (38.4%) have both out-edges and in-edges. About 50,609 protein sequences (5.2%) only have out-edges; while 424,765 protein sequences (43.6%) only have in-edges. The average degree of this graph was 23, with an average out-degree of 46 and an average in-degree of 24.5. We note that traditional ACPM algorithms, such as those of Weiner et al and Uliel et al,22,23,53,54 which do not consider the all-against-all problem, will find CPs for only the 374,279 sequences that have both in-edges and out-edges.
Figure 5A shows the number of directly connected pairs in the Top

Number of directly connected pairs in Top
Predicting functions for uncharacterized proteins
We first tested the function prediction using nine multidomain proteins in the ProDom dataset, with known functions in GO. Table 3 shows the prediction results on nine sample multidomain proteins using the union of the functions from the proteins in the in-edge and out-edge sets at different thresholds on the
Predicted functions for nine sample multidomain proteins using the union of functions for known proteins in the in-edge and out-edge sets.

Predicted functions for nine sample multidomain proteins using the intersection of functions for known proteins in the in-edge and out-edge sets.

We conducted a larger experiment to predict the protein functions in the Top 500 highest degree proteins in our network. Of these, 156 proteins were not found in the GO database. Thus, performance analysis was based on the remaining 344 multidomain proteins that have function annotation in GO. Prediction performance was measured in terms of
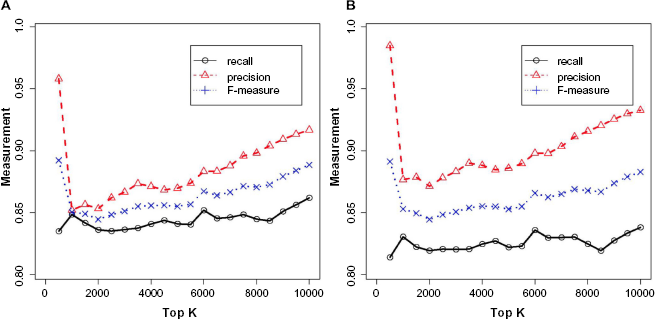
Performance in function prediction based on circular permutations for the top
Predicting novel multidomain proteins associated with cancer
In this experiment, we use the proposed multi-domain protein circular relationship network, and tailor the function prediction method described, to specifically focus on prediction of novel multidomain proteins with potential associations with cancer. Using data from the Cancer Resource dataset, 63 we selected five types of cancer (bone, colon, lung, skin, and breast cancers), and studied subnetworks involving multidomain proteins known to be associated with each type of cancer.
Construction of cancer subnetworks
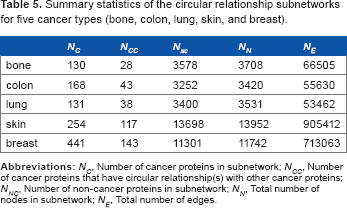
For each cancer type, we construct a corresponding subnetwork using only the multidomain proteins identified in the Cancer Resource dataset. Thus, we determine the proteins that have circular relationship(s) with other proteins involved in the same cancer type. For instance, this yields 28 proteins for bone cancer, and 43 proteins for colon cancer. Using these known cancer proteins that are associated by CPs, we then search the larger circular relationship network with all the multidomain proteins in the ProDom database. We thus obtain a larger subnetwork, whereby nodes in the subnetwork are proteins with known association to a given cancer type, or those that are associated with these through a CP relationship. Figures 7 and 8 show the subnetworks for colon and skin cancers, respectively. The subnetworks for colon, lung, and breast cancers are available as supplementary material. Table 5 shows the summary statistics of the subnetworks from the circular relationship network, for each of the five cancer types.

The subnetwork of colon cancer proteins. Red nodes denote known cancer proteins; yellow nodes are the proteins predicted to be associated with colon cancer.
Summary statistics of the circular relationship subnetworks for five cancer types (bone, colon, lung, skin, and breast).
Predicting cancer-related multidomain proteins
Using the cancer-type specific subnetworks, we can now predict which multidomain proteins are most likely to be associated with the given cancer type. This requires only a slight modification of the basic function prediction method described earlier in the Materials and Methods section.
We still use the notion that hubs (nodes with a higher connectivity) are more likely to be important in the subnets. That is, these nodes have more circular relationships in this cancer protein subnetwork.
However, rather than using simple connectivity based on node degree distributions, we measure the node
In this work, we consider the Top 10 multidomain proteins in the betweeness centrality ranking as predicted to be associated with the given cancer type, and then use literature search to further validate the predicted associations. See Figures 7 and 8, and figures in Supplementary Material. In the figures, red nodes denote known cancer proteins for the given cancer type, yellow nodes are the predicted proteins (those with largest betweenness centrality values in the subnetwork). Observe that the yellow nodes tend to have much more connections in the network, and they tend to be linking different regions in the network, showing their significance.

The subnetwork of skin cancer proteins, showing only the Top 200 nodes (ranked by betweenness centrality).
Table 6 lists the 10 proteins with the highest betweenness centrality measures in each of the five cancer subnetworks. For each cancer type, these proteins are the most important nodes, with respect to CP relationships, and usually have more connection with the cancer proteins.
Top 10 proteins with the highest betweenness centrality values in each of the five cancer subnetworks (bone, colon, lung, skin, and breast cancer).

indicates that the protein was in the list of known cancer proteins from the Cancer Resource dataset.
Literature search
Given the cost and expense of wet-lab experimental verification, it is important to further narrow down the list of predicted proteins. For this purpose, we use literature search on PubMed to determine whether there has been previous publications on the predicted connection between the multidomain protein and the given cancer type. We also use information from the Atlas of Genetics and Cytogenetics in Oncology and Haematology (http://atlasgeneticsoncology.org). If a protein is already listed in the Atlas, we assume that it is known to be related with cancer. In some cases, the relationship may not be with the specific cancer type that our system predicted. Thus, for literature-based validation, we searched for the Top 10 predicted proteins for each cancer type, shown in Table 6. If the predicted association has not been previously reported in PubMed, or not in the Atlas, we take it to be a novel association found by our method.
From the table, we can observe that several of the Top 10 proteins for a given cancer type also appeared in the Top 10 for other cancer types (eg, PAK7 and stk-42 shared by bone and colon; HTK16, HTK98, pik3r1, and ced-2 shared by bone and lung). This is not completely unexpected, given that certain proteins are known to be implicated in multiple cancer types (see also the column labeled “In CT”). Lung cancer shared more circular proteins in the Top 10 with bone cancer. More significantly, some of the multidomain proteins in the Top 10 have previously been reported in PubMed or in the Atlas to be involved in some cancers, but not necessarily in the cancer type that was predicted by the proposed method. Of the Top 10 proteins predicted for each cancer type, we have the following known associations (from Cancer Resource) or reported associations (in PubMed) with the specific cancer type predicted: bone 2, colon 1, lung 2, breast 4, and skin 2. This means that most of the predicted proteins have not yet been connected with the specific cancer predicted. The table also shows the predicted proteins that are described in Uniprot as uncharacterized, unknown, putative, probable, or hypothetical (denoted as U). Those with U* are assumed to be known, since they have already been included in the Cancer Resource dataset. 63 In fact, a good number of the predicted proteins have not been previously reported to be connected with any cancer type in the literature: bone 3, colon 6, lung 3, breast 5, and skin 3 (not including the deleted (obsolete) proteins). These proteins, along with those denoted with a U in the table, represent a reduced set of multidomain proteins that are most likely associated with the specified cancer types, and can thus be subjected to further wet-lab verification.
One striking observation from the table is the fact that all the Top 10 proteins for skin cancer have the same value of 49 for

A 49-node dense subgraph at the center of the skin cancer subnetwork. See also Figure 8.
Discussion and Conclusion
We identify three major contributions of this work. First, we proposed an efficient algorithm for rapid identification of both exact and approximate CPs in multidomain proteins. By analyzing the computational complexity of the algorithms, we showed their superiority over current state of the art. We also presented results on the practical running time required by the algorithms. Second, we showed how the circular relations can be used to construct a network between these proteins, based on which we perform functional annotation of multidomain proteins. This method showed a performance of about 0.81 precision and 0.88 recall, using known functions from GO on the Top 500 proteins. Third, we extended the method to construct subnetworks for selected cancer subtypes and performed prediction of the association between multidomain proteins and the selected cancer types. Our prediction based on the Top 10 proteins with the highest betweeness centrality measures contained many uncharacterized multidomain proteins that are likely to be associated with specific cancer types. Some of the multidomain proteins are predicted to be associated with more than one cancer type.
Of note is the observed 49-protein dense subgraph for the skin cancer subnetwork, which contains the top-ranking proteins predicted to be associated with skin cancer. We are not aware of any previous report of such a dense subgraph of multidomain proteins related by CPs, with known associations to skin cancer. Thus, we do not have a hard evidence on the practical relevance of the observed dense network to skin cancer. However, within this subgraph, we can identify groups of genes that are implicated in various functions that are relevant to cancer. For instance, we can observe several known cancer-related functional groups: Pro-oncogenes and growth-promoter genes 72 (Ntrk3, Stk25, flt1, Cdk3); genes involved in inducing angiogenesis 73 (EIF2AK2, Ntrk3, Stk25, flt1); genes for regulating oxidative stress 74 (Bcpk1, hog1); genes for regulating protein phosphorylation and dephosphorylation75,76 (NEK3, Stk25, Cdkl2, Stk25, Cdk3, CIPK9, HIPK4, EIF2AK2); gene for regulating glucose level (SPS1); genes for regulating cell cycle, cell migration, and metastasis76–79 (FGL1, flt1, nek1, CDC2, hog1, Cdk3, EIF2AK2); and genes for other functions. This long list of cancer-related genes in the 49-node dense subgraph gives us some confidence on the relevance of this network. We can also expect that some of the unknown/uncharacterized proteins in this 49-node dense network are likely to be implicated in some cancers, especially in skin cancer. It will be interesting to study this quasi-clique further, for any biological relevance to molecular studies of skin cancer in particular and of cancer in general.
We note that our prediction for associations with cancer is based primarily on information from Cancer Resource data-set. 63 Thus, a multidomain protein that is not in the list will be predicted as a potentially novel association with the given cancer type. This might explain why some of the predicted associations are already observed in the Atlas or in PubMed. Yet, this still gives some credence to the power of the proposed method: it can find important associations between cancer-related proteins. Verifying whether the association is novel or not can be performed easily.
Our approach is a computational method, which essentially generates hypotheses on potential functional associations for multidomain proteins. This provides an important mechanism needed to prune down the large number of possibilities for later biological verification of the predicted associations in the wet-laboratory.
Author Contributions
Conceived and designed the experiments: DA, JL, YJ. Analyzed the data: DA, JL, YJ, BHJ. Wrote the first draft of the manuscript: DA, JL. Contributed to the writing of the manuscript: DA, JL, YJ, BHJ. Agree with manuscript results and conclusions: DA, JL, YJ, BHJ. Jointly developed the structure and arguments for the paper: DA, JL, YJ, BHJ. Made critical revisions and approved final version: DA, JL, YJ, BHJ. All authors reviewed and approved of the final manuscript.