
Abstract
The advance in human genome sequencing technology has significantly reduced the cost of data generation and overwhelms the computing capability of sequence analysis. Efficiency, efficacy, and scalability remain challenging in sequence alignment, which is an important and foundational operation for genome data analysis. In this paper, we propose a two-stage approach to tackle this problem. In the preprocessing step, we match blocks of reference and target sequences based on the similarities between their empirical transition probability distributions using belief propagation. We then conduct a refined match using our recently published sparse-coding belief propagation (SCoBeP) technique. Our experimental results demonstrated robustness in nucleotide sequence alignment, and our results are competitive to those of the SOAP aligner and the BWA algorithm. Moreover, compared to SCoBeP alignment, the proposed technique can handle sequences of much longer lengths.
Keywords
Introduction
In bioinformatics, sequence alignment is an important way to identify similar regions that might be associated with similar functional and structural relationship between sequences. With the quick growth of genomic data, it is important to develop effective sequence alignment techniques that are scalable. The past decade has witnessed the development of many sequence alignment technologies. Cancers are caused by the collection of genomic sequence changes. 1 Therefore, alignment and analyses of cancer genome sequences provide basics to understand cancer biology, diagnosis, and therapy.
In general, pairwise sequence alignment methods can be classified into local and global approaches. The global alignment attempts to find the best match between two strings with similar lengths through global optimization. In contrast, the local alignment is usually used to identify regions of similarity between a short query and a longer sequence. Global alignments2–5 are less prone to demonstrating false homo logy as each letter of one sequence is constrained to being aligned to only one letter of the other. Local alignments,6–9 on the other hand, can cope with rearrangements between non-syntenic, orthologous sequences by identifying similar regions in sequences; this, however, comes at the expense of a higher false positive rate because of the inability of local aligners to take into account overall conservation maps. 10
A lot of efforts have been made to improve the efficiency and efficacy of sequence alignments. The ClustalW program proposed by Thompson and Larkin11,12 uses a multi-stage mechanism to weigh and to align subsequences based on sequence divergences. In addition, sequence annealing technique incrementally builds sequence alignment one at a time by checking whether a single match is consistent with a partial multiple alignment. 13 Darling et al proposed a hidden Markov model that uses a sum-of-pairs breakpoint score to facilitate the detection of rearrangement breakpoints, when genomes have unequal gene content. 14 Mummer is a highly efficient suffix tree-based matching tool for whole genome alignment as well as incomplete genomes. 15
Researchers also proposed heuristics to accelerate sequence alignment. For example, the bounded sparse dynamic programming (BSDP) is used to support rapid approximation of exhaustive alignment in Slater and Birney. 16 Another heuristic-driven approach, namely FastTree, is a tree-based method that stores profiles of internal nodes in a tree, such that candidate joins can be quickly identified. FastTree is also scalable for hand ling alignments over 10,000 sequences.17,18
Maximum-likelihood-based approaches like PhyML and RAxML-VI-HPC have been developed as well. PhyML 19 used a hill-climbing algorithm that adjusts tree topology and branch length at each tree modification iteration. RAxML-VI-HPC, 20 which stands for randomized accelerated maximum likelihood for high-performance computing, takes advantages of a parallel program to support large-scale genome alignment.
In this paper, we propose a novel alignment method that uses sparse coding 21 and empirical transition probability to tackle the scalability challenge. Tanks to the sparse representation, our mechanism can handle long sequences with reduced memory footprint. We also leverage belief propagation (BP) to combine local and neighboring information of candidate nucleotides into consideration, and generate matching scores to determine the best match. The rest of this paper is structured as follows. Section II introduces our proposed method. Section III presents our results, including the comparison against SOAP aligner 22 and BWA. 23 Finally, we draw our conclusions in Section IV.
Proposed Method
In this section, we present our genome indexing and alignment framework in detail, where the proposed method includes three steps: indexing, index matching, and sequence matching. In this paper, we refer to “reference sequence” as the baseline sequence and try to align a “read sequence” against the baseline sequence.
Indexing
The current genome indexing methods generate huge indices before performing the actual alignment to decrease the alignment time.24,25 The indexing process can be very time-consuming. In contrast, our proposed indexing technique provides a faster and light-weight alternative for index generation, which is similar to the big data retrieval systems that were proposed.26–28 These indices can reduce the search space and provide an estimation of the read sequence locations in the reference sequence. The proposed genome indexing technique models a nucleotide sequence as a graph by counting the transitions between each pair of nucleotides. To be more specific, as shown in Figure 1, we consider a graph with four states according to the different types of nucleotides and 16 vertices according to all possible transitions between nucleotides. We read the first nucleotide of the sequence and treat it as the initial state. Then, we move from one state to the other state by scanning the next nucleotide repeatedly till the end of the sequence. Afterward, we calculate the number of nucleotide transitions where we count how many times we pass one vertex in the graph and store them in a 4 x 4 matrix. Finally, we normalize the resulting matrix as follows:

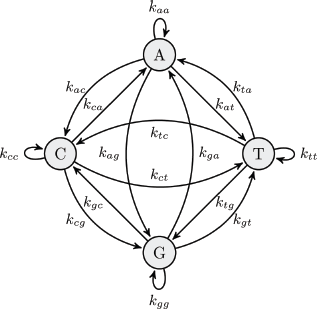
The transition diagram between nucleotides.
If the length of a sequence is larger than a given threshold, ie,

An example of the indexing procedure for a small sample subsequence.
Index matching
The index matching step is designed to find similar indices based on global information of the sequence. We define a symmetric distance function between two index matrices,
After generating the indices of the reference sequence and the read sequence, the
We apply BP to the factor graph of the test sequence with
Then, the candidate index numbers are fed to a factor graph, and the corresponding
Sequence matching
The sequence matching step is based on sparse coding and BP algorithm. In this step, we use the subsequences that were selected in the previous step to generate an over-complete dictionary. Then, for each nucleotide in the read sequence, we pick
Implementation details

[
Proposed nucleotide sequence alignment algorithm for estimating the location of the input sequence
We designed our experiments based on the work in Darling et al. 14 to evaluate the proposed method for aligning the nucleotide sequences and to compare it with SOAP aligner, 22 BWA, 23 and 1D-SCoBeP 31 We considered the problem of aligning a sequence of human nucleotides from the National Center for Biotechnology Information 34 and Cancer Genomics Hub. 35
To evaluate the performance of our approach, we conducted two sets of tests on the nucleotide sequences. In the first set, we selected 50 short subsequences of human genomes and then used SOAP aligner, BWA, 1D-SCoBeP, and the proposed method to find the location of selected subsequence nucleotide in the human chromosome. All the four algorithms successfully passed this test. We created 20 shuffled subsequences of the reference sequence as follows: for each read sequence
Figure 3 shows that the result of the 1D-SCoBeP and the proposed method show a better performance with a gap of 100–120 nucleotides away from the ground truth. Since we were using non-overlapped subsequences for the dictionary generation, the gap between the proposed method and the ground truth was larger than those reported in 1D-SCoBeP.
31
In our experiments, the following parameters were used: the number of candidate points

The results of proposed method for non-collinear nucleotide sequence alignment are shown. (
To evaluate the robustness of the proposed method, we generate indices for long human genome sequences (ie, 5 x 108 nucleotides), where
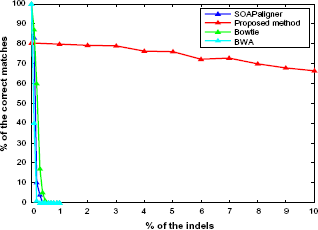
We investigate the impact of small indel rate in the range from 0.5 to 1.5% in Figure 5. In this figure, we showed the accuracy of 1% indels in red for the dataset used in Figure 4 as reference. To verify our result, we repeat the experiments with different indel steps and different read locations, and present the results in green and blue, respectively. Note that each point in this Figure was obtained from the evaluation over 105 read sequences. There are slight variations among the curves because of statistical deviation. The summary of the indel rate accuracy is shown in Table 1.

The percentage of successful alignments in the presence of 0.5–1.5% indels. The green line is the percentage of successful alignments, where the rate of the indels is changing between 0.7 and 1.3% at equal step of 0.05%. The blue line is the percentage of successful alignments, where the rate of the indels is changing between 0.5 and 1.5% at equal step of 0.1%, and the red line is the same as in Figure 4. Each point represents 105 random site selection with same indel rate. Note that the genome sequences used in this study were obtained from the National Center for Biotechnology Information and the Cancer Genomics Hub.34,35
Percentage of successful alignments.

The computational complexity of proposed is mainly determined by the following five steps: (1) indexing, (2) index matching, (3) extracting subsequence nucleotides as features and constructing the dictionary, (4) finding candidate nucleotides via sparse coding, and (5) applying BP. Assume that the sizes of the read and reference sequences are
In this paper, we proposed a sparse coding and BP-based method for indexing and alignment genome sequences. The proposed method builds a transition matrix based on the neighboring nucleotides of an input sequence and then reduces the search space by selecting the top
Author Contributions
Conceived and designed the experiments: AR, NB, SW, XJ, SC. Analyzed the data: AR, NB, SW, XJ, SC. Wrote the first draft of the manuscript: AR, NB, SW, XJ, SC. Contributed to the writing of the manuscript: AR, NB, SW, XJ, SO Agree with manuscript results and conclusions: AR, NB, SW, XJ, SO Jointly developed the structure and arguments for the paper: AR, NB, SW, XJ, SO Made critical revisions and approved final version: AR, NB, SW, XJ, SO All authors reviewed and approved of the final manuscript.