
Abstract
Cancer risk prediction models are important in identifying individuals at high risk of developing cancer, which could result in targeted screening and interventions to maximize the treatment benefit and minimize the burden of cancer. The cancer-associated genetic variants identified in genome-wide or candidate gene association studies have been shown to collectively enhance cancer risk prediction, improve our understanding of carcinogenesis, and possibly result in the development of targeted treatments for patients. In this article, we review the cancer risk prediction models that have been developed for popular cancers and assess their applicability, strengths, and weaknesses. We also discuss the factors to be considered for future development and improvement of models for cancer risk prediction.
Introduction
Cancer is one of the leading causes of death worldwide. A large percentage of patients are diagnosed at an advanced stage, making the removal of tumors in this population problematic. As a result, the overall 5-year survival rate is low for this cohort of patients. 1 Therefore, early stage detection would be helpful in reducing cancer mortality because treatment might be most effective at the earliest stages of the disease. For this reason, a well-established assessment model would greatly benefit patients, clinicians, and researchers because it would allow individuals at high risk to be identified at the earliest stages.
Cancer is a polygenic disease in which many genetic factors appear to play important roles in disease development in its different subtypes of cancer.
2
To date, more than 50 cancer genome-wide association studies (GWAS) incorporating more than 15 different malignancies have been reported identifying over 100 genomic cancer susceptibility regions.
3
The cancer-associated genetic variants identified in GWAS or candidate gene association studies have been shown to collectively enhance cancer risk prediction, improve our understanding of carcinogenesis, and possibly result in the development of targeted treatments for patients. For example, clinicians already use these kinds of guidelines in making decisions about assessments in order to identify carriers of
Breast Cancer
Although the incidence rate of breast cancer has been declining since 1998–1999, there will still be 232,670 new female cases and 2,360 new male cases in the US in 2014 (http://www.cancer.gov/cancertopics/types/breast). Early stage detection of breast cancer is very important because treatment can be more effective at the early stages. For this reason, a well-established assessment model that could identify individuals at high risk would greatly benefit patients, clinicians, and researchers in the prevention and intervention of breast cancer.
Risk prediction models have been widely used to identify individuals with high risk of breast cancer. The Gail model,5,6 for example, is used by FDA to screen women with high risk for chemopreventive use of tamoxifen. Traditional risk factors like family history, age at menarche, age at first live birth and number of previous breast biopsies, mammographic density, and diagnosis of atypical hyperplasia have been used to predict breast cancer as well.
Recently, genetic susceptibility risk prediction has been improved with the discovery of more than 40 risk-associated single-nucleotide polymorphisms (SNPs) from GWAS. Several breast cancer susceptibility genes have now been identified, including
Wacholder et al. 7 evaluated the Gail model using 5,590 cases and 5,998 control subjects, which are from four US cohort studies as well as from a Poland case control study. The range of age of all the subjects is from 50- to 79-years old. The area under the receiver-operating-characteristic (ROC) curve (AUC) ** is 58% for four traditional risk factors. After incorporating 10 genetic variants into the prediction using a logistic regression model, the Wacholder study achieved a 61.8% AUC, a 3.8% increase over the model without genetic variants. Another notable study in breast cancer risk prediction was done by Machiela et al. 8 In this study, a total of 1,145 breast cancer cases and 1,142 controls from the Nurses’ Health Study were used to build and evaluate polygenic risk scores (PRSs) with 10–60,000 independent SNPs showing the strongest evidence of association with breast cancer. No significant evidence was found that polygenic risk score (PRS) using common variants could improve risk prediction for breast cancer over replicated SNP scores that had been robustly replicated across several independent sample sets.
Some polymorphisms identified in GWAS were also associated with an increased risk of breast cancer for
Compared to high-penetrance mutations, such as
Appendix B. Description for the Abbreviations of Gene Names in this Article.
More details about basic concepts used in this article can be found in Appendix A.
Prostate Cancer
Prostate cancer, behind only lung cancer, is the second leading cause of cancer-related deaths in American men. Recent data indicate that the estimated probability of being diagnosed with prostate cancer is 2.5%, 7%, and 13% for men ages 40–59 years, 60–69 years, and 70 years and older, respectively. In 2014, 233,000 new cases will be diagnosed in the US, and more than 29,480 men die of the disease (http://www.cancer.org/cancer/prostatecancer/detailedguide/prostate-cancer-key-statistics).
Prostate cancer is also a complex and unpredictable disease, with the risk for cancer affected by advancing age, ethnic background, and family history. 12
Prostate cancer is usually accompanied by a rise in the concentration of serum prostate-specific antigen (PSA). PSA lacks specificity, but, nevertheless, has been used for decades as a sensitive biomarker and has evolved into a controversial predictor of prostate cancer mortality. In general, prostatic biopsies are often deemed unnecessary, which underscores the need for improving prediction models with increased specificity in order to aid clinicians when deciding whether or not to recommend a biopsy for patients. This is especially relevant for men with mildly elevated PSA values (3–10 ng/mL), where the risk of being diagnosed with prostate cancer is only about 20–25%. 13 After diagnosis, some cancers are indolent and cause no clinical problems, whereas others progress and may become fatal. Therefore, it is important to search for biomarkers that signal a need for more aggressive treatments, potentially improving clinical outcomes. Recently, more than 30 discovered SNPs have been associated with prostate cancer. 14 These SNPs provide an opportunity to identify strong candidates for a predictive role. SNPs identified and associated with prostate cancer in GWAS are common but confer only small increases in the risk. The mechanisms underlying their association with prostate cancer risk remain unknown.
Xu et al. 15 used SNPs of multiple DNA sequence variants and family history to estimate the absolute risk for prostate cancer. These investigators examined a Swedish study with 2,893 cases and 1,781 controls and a study in the US - the Prostate, Lung, Colon and Ovarian Cancer Screening Trial with 1,172 cases and 1,157 controls. Individuals with more than 14 risk alleles and positive family history had almost a five-fold increase in risk compared with people who had 11 risk alleles and negative family history. The study also outlined the risk of developing prostate cancer for a 55-year-old man who has positive family history and more than 14 risk alleles as being 40% over the next 20 years, while men without family history and such genotypes saw their absolute risk reduced to 13%.
In another study by Sun et al. 16 the investigators assessed predictive performance by employing positive predictive values (PPV) as well as sensitivity using family history and three sets of SNPs associated with prostate cancer. This study was a population-based case-control study (2,899 cases and 1,722 controls) in another Swedish population. SNPs and family history emerged as factors that can differentiate individual risk for prostate cancer, while identifying men at higher risk. In this particular study, the top 18% of men had a two-fold risk, while the top 8% had developed a three-fold risk of having prostate cancer in a 20-year period (age range: 55–74 years). In addition, the study showed that including more SNPs in the risk prediction will increase sensitivity with PPV.
Other studies have combined genetic variants along with PSA to predict the risk of prostate cancer. For example, a study by Johansson et al. 17 specifically combined 33 genetic variants with PSA and evaluated the risk. This was a case control study (520 cases and 988 controls) nested within the Northern Sweden Health and Disease Cohort. The AUC was used to assess whether the GRS of 33 SNPs in addition to pre-diagnostic PSA improves prostate cancer prediction. Adding GRS into the model improved the AUC from 86.2% to 87.2%. Thus, it appears that including GRSs into these models may not be beneficial when competing for a clinical risk assessment of prostate cancer.
Others such as Machiela et al. 8 created a new model by applying PRSs 18 and incorporating common variants to predict the risk of prostate cancer. A total of 1,164 prostate cancer cases and 1,113 controls from the Prostate Lung Colorectal and Ovarian Cancer Screening Trial were employed in this study. PRSs with 10 to 60,000 independent SNPs were used in the PRS model to compare with a model only including 30 published risk variants. Appling a 10-fold cross-validation for PRS model, the area under the ROC curve ranged from 0.564 (60,000 SNPs) to 0.569 (10 SNPs), while the AUC using 30 published risk SNPs from the literature was 0.614. Kote-Jarai 19 also proposed a study to predict the risk of prostate cancer using a multiplicative risk model, which combines the risk variants. This study used a worldwide consortium, composed of 13 groups with 7,370 prostate cases and 5,742 controls. All of the loci contributed to 16% of the familial risk of the disease, and the top 10% of risk distribution doubled the chance of prostate cancer with an odds ratio of 2.1.
The first risk prediction model for familial prostate cancer was developed by Macinnis et al. 20 which incorporated 26 prostate cancer-associated SNPs identified in previous GWAS. 21 Family phenotypes and histories were explained by a mixed model of inheritance which can be used to predict the probability of developing prostate cancer for an individual. Combined populations from 1,832 prostate cancer patients and relatives in Australian and 2,558 patients from prostate cancer clinics at the Royal Marsden NHS Foundation Trust (UK) were used. Using this predictive model, the risk of prostate cancer for an UK male can be predicted. For example, if a man's genotype is in the top 10th percentile of joint genotype distribution and his father was diagnosed with prostate cancer at age 70, he would have a cumulative risk of 33% of developing prostate cancer by age 85. For a male with a genotype risk within the bottom 10%, the risk to develop prostate cancer would be 23%. In comparison, even without SNP information and incorporation into this kind of model, the risk remains 22% for a UK man.
Finally, Lindström et al. 22 combined a series of risk models and estimated their performance in 7,509 prostate cancer cases and 7,652 controls within the National Cancer Institute Breast and Prostate Cancer Cohort Consortium. The investigators also calculated absolute risks based on the Surveillance, Epidemiology, and End Results incidence data. The best risk model included individual genetic markers and family history of prostate cancer. They observed a decreasing trend in discriminative ability with advancing age, with highest accuracy in men younger than 60 years. The absolute 10-year risk for 50-year-old men with a family history ranged from 1.6% (10th percentile of genetic risk) to 6.7% (90th percentile of genetic risk). For men without a family history, the risk ranged from 0.8% (10th percentile of genetic risk) to 3.4% (90th percentile of genetic risk). These results indicate that incorporating both genetic information and family history into prostate cancer risk models can be particularly useful for identifying younger men who might benefit from PSA screening.
Testicular Cancer
Testicular cancer remains the most common form of cancer in men between the ages of 15 and 35 (http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0002266/). It is also the most treatable form of cancer with a survival rate greater than 95% for the least aggressive type.
The risk of this type of cancer has been reported to be 8- to 10-fold higher for brothers and two- to four-fold higher for the sons of men who previously had testicular cancer.23–27 Familial studies have estimated that genetic effects account for nearly a quarter of testicular cancer risk, which is one of the largest estimated heritabilities reported for any type of cancer.
28
More specifically, GWASs have implicated multiple genomic regions associated with testicular cancer risk, including those containing
Previously published GWAS and candidate gene data have also been used to build a multiplicative model with risk variants and estimate the AUC as a measure of discrimination between testicular cancer cases and controls. Kratz et al.'s
34
study is one such example, of using this kind of data,29–33 where previously uncovered predisposition alleles in or near
We have to be aware as well that white men are more likely to develop testicular cancer than African-American and Asian-American men. Because of this race/ethnicity effect, cancer risk prediction needs to be tailored for specific populations. Additionally, GWAS needs to be extended into different populations. Each specific population requires models developed with their specificity in mind in order to create improved methods for overall risk assessment of testicular cancer.
Lung Cancer
Lung cancer remains the most common form of human cancer with complex risk factors, including genetic and environmental effects. Heredity plays an important role, and in relatives of people with lung cancer, the risk is increased 2.4 times.36,37 This may be due, for example, to risks associated with genetic polymorphisms.38–43 Environmental factors, such as a history of smoking, are central to several proposed lung cancer risk assessment models. These models include the Bach model, Spize model, and Liverpool Lung Project (LLP) model44–47 as well as the improvement models based on LLP.48,49 The LLP risk model, 45 developed from the LLP case-control study, provides a single unified model for smokers (current and former) and nonsmokers, whereas the Bach model was developed for predicting risk only in smokers and the Spitz model 46 requires three separate models for predicting risk in current smokers, former smokers, or nonsmokers. In addition, the LLP model also accounts for important lung cancer risk factors in addition to age, sex, and smoking duration. These include history of pneumonia, a history of non-lung cancer, prior asbestos exposure, and family history. Overall, this comprehensive model is simpler to incorporate into a clinical setting than Tammemagi and colleagues’ model, 47 which includes many smoking-related variables that may be difficult to obtain from patients during clinical exchanges.
Other models have been developed in order to predict the 5-year absolute risk of lung cancer. For example, model based on five epidemiologic risk factors has been developed by the LLP by Raji et al. 48 where investigators quantified the improvement in risk prediction with the addition of SEZ6L, a Met430IIe polymorphic variant linked with an increased risk of lung cancer, within the framework of the LLP risk model. In this predictive model, the authors combined the genotypes of 388 LLP subjects on SEZ6L SNP with epidemiologic risk factors. They use multivariable conditional logistic regression, with and without SEZ6L SNP, to predict 5-year absolute risk of lung cancer. Pair-wise comparison of the AUC and the net reclassification improvements (NRI) were also used to assess the improvement in the model itself with and without the SEZ6L SNP. The authors found a modest statistically significant increase in AUC when SEZ6L was added into the baseline model. The NRI for the genetic model was 27% with the SNP, while 15% without the SNP.
Raji et al. also further evaluated the LLP risk model in terms of discrimination and its ability to demonstrate a predictive benefit for stratifying patients for computed tomography (CT) screening. 49 These investigators assessed the 5-year absolute risks for lung cancer that were predicted by the LLP model in both case-control and prospective cohort study, which used data from three independent studies – the European Early Lung Cancer (EUELC), Harvard case-control studies, and the LLP population-based prospective cohort (LLPC) study from Europe and North America. The LLP risk model produced good discrimination in both the Harvard (AUC = 0.76 [95% confidence interval, CI, 0.75–0.78]) and the LLPC (AUC, 0.82 [CI, 0.80–0.85]) studies and modest discrimination in the EUELC (AUC, 0.67 [CI, 0.64–0.69]) study. The decision utility analysis, which incorporates the harm and benefit of using a risk model to make clinical decisions, indicated that the LLP risk model performed better than smoking duration or family history alone in stratifying high-risk patients for lung cancer CT screening. However, this model cannot assess whether the incorporation of other risk factors, such as lung function or genetic markers, will improve accuracy. In particular, the lack of information on asbestos exposure in the LLPC limited the ability to validate the complete LLP risk model.
Models and risk evaluation focused on genetic susceptibility loci have conferred a small to moderate disease risk and appear to be of limited utility in risk prediction. Li et al.
50
combined multiple disease-related loci with modest effects into a GRS and identified subgroups that were at high risk of lung cancer in a Chinese population. In their case-control study, they evaluated the discriminatory and predictive ability of the cumulative effect of several SNPs associated with lung cancer risk. Five SNPs identified in previous GWA or large cohort studies were genotyped in 5,068 Chinese case-control subjects. The GRS based on these SNPs was estimated by two approaches: a simple risk alleles count (cGRS) and a weighted (wGRS) method. The AUC in combination with the bootstrap resampling method was used to assess the predictive performance of the GRS for lung cancer. Four independent SNPs were found to be associated with a risk of lung cancer. The wGRS based on these four SNPs was a better predictor than cGRS. Using a liability threshold model, they estimated that these four SNPs accounted for only 4.02% of genetic variance in lung cancer. As with other studies, smoking history contributed significantly to lung cancer risk (
Black/white disparities concerning in lung cancer incidence and mortality mandate an evaluation of underlying biological differences. Etzel et al.
51
have previously shown higher risks of lung cancer associated with prior emphysema in African-American populations compared with white patients with lung cancer. Spitz et al.
52
further evaluated a panel of 1,440 inflammatory gene variants in a two-phase analysis (discovery and replication), adding top GWAS lung cancer hits from white populations, and 28 SNPs from a published gene panel. The discovery set (477 self-designated African-Americans cases, 366 controls matched on age, ethnicity, and gender) was from Houston, Texas. The external replication set (330 cases and 342 controls) was from the EXHALE study at Wayne State University. In discovery, 154 inflammation SNPs were significant (
Bladder Cancer
Bladder cancer remains a major health issue worldwide. In the US, bladder cancer is the fourth most common tumor in men and an estimated 74,690 new diagnoses are expected in 2014 (http://www.cancer.org/cancer/bladdercancer/detailedguide/bladder-cancer-key-statistics). The disease generally presents in older individuals, and is more common in men than women, with higher frequency among white patients than those of other ethnicities. Smoking is the most widely recognized cause of bladder cancer and accounts for half of all cases in the US.
The first risk prediction model for bladder cancer was developed by Wu et al. 53 in 2007. Patient epidemiologic and genetic data from a case-control study were used to build risk prediction models and constructed ROC. The AUC was used to evaluate the model's discriminatory ability. The model consisted of 678 white patients and 678 controls and included mutagen sensitivity and pack-years as well as six other risk factors, while achieving a 0.80 AUC, demonstrating good discrimination ability. In 2009, 54 the same group added three bladder cancer predisposition SNPs into the risk prediction model but found no improvement of the discrimination power. However, Chen and colleagues 54 also pointed out that with the development of computing power and statistical tools, other risk factors such as gene–gene interaction and gene-environment interactions may offer greatly improved risk prediction.
In a recent paper published in
Head and Neck Cancer
The incidence of head and neck cancer has increased markedly in the last 20 years. Head and neck cancers account for about 3–5% of all cancers in the US. In this year, an estimated 55,070 people (40,220 men and 14,850 women) will develop head and neck cancers and 12,000 deaths (8,600 men and 3,400 women) will occur (http://www.cancer.net/cancer-types/head-and-neck-cancer/statistics).
Cigarette smoking is associated with increased head and neck cancer risk and tobacco-related carcinogens are known to cause bulky DNA adducts. Nucleotide excision repair genes encode enzymes that remove adducts and may be independently associated with head and neck cancer risk, as well as modifiers of the association between smoking and head and neck cancer risk.56–58
Several studies have reported that SNPs of genes in multiple biological pathways are involved in the development of head and neck cancer.59–63 Recently, Annah et al. 61 performed a two-stage GWAS with a total of 8,605 cases and 11,405 controls and reported that five genetic variants had significant associations with risk of upper aerodigestive tract cancers including head and neck cancer in Europeans.
With the recent increase in associated SNPs with head and neck cancer being identified, the development of risk prediction models is catching up. A study by Wu et al. 64 used a customized chip containing 9,645 chromosomal and mitochondrial SNPs (mtSNPs) to call genotypes for 150 early stage head and neck cancer patients with 300 controls. The goal is to model the second primary tumor or head and neck cancer recurrence using both clinical and epidemiological variables. Results showed that when 12 chromosomal SNPs and one mtSNP were incorporated into the model, the AUC increased from 0.64 to 0.84. The 95% CI of the AUC difference is 0.18–0.29, indicating significant improvement in discrimination power.
Discussion
Cancer is a polygenic disease in which many genetic factors appear to play important roles in disease development in its different subtypes of cancer. 2 During the past several years, more than 100 SNPs have been identified that are associated with cancer. 3
How to effectively incorporate these genetic susceptive variants in risk predictive models has become more and more important during the clinical decision-making process because effective models can help physicians and patients determine whether a genetic testing is needed. Although enormous progress has been made in the area of genetics and the susceptive risk prediction of cancer, cautions should be made when considering the application of these models within the clinical setting. Cancer remains a fundamentally complex disease with multiple, interacting risk factors. These risk factors include components of race/ethnicity, environmental carcinogens, familial history, genetic variants, and their interactions. The studies reviewed here should be understood as an initial attempt to begin a more systematic approach to assessing predictive risk models for cancer treatment in the future. In order to more accurately predict the overall risk of cancer in patients, risk prediction models need to be continuously reexamined, comprehensively assessed, and revised, taking into consideration specific populations and emergent subtype of cancer.
Table 1 indicates that the cancer risk prediction models with genetic variants generally outperform the models without genetic variants in both discrimination and prediction of cancer. However, there are still many practical concerns on implementing genetic testing into the diagnostic process. For example, the substantial cost of genetic screening is one of the main concerns.
Performance of cancer risk prediction models with genetic variants.
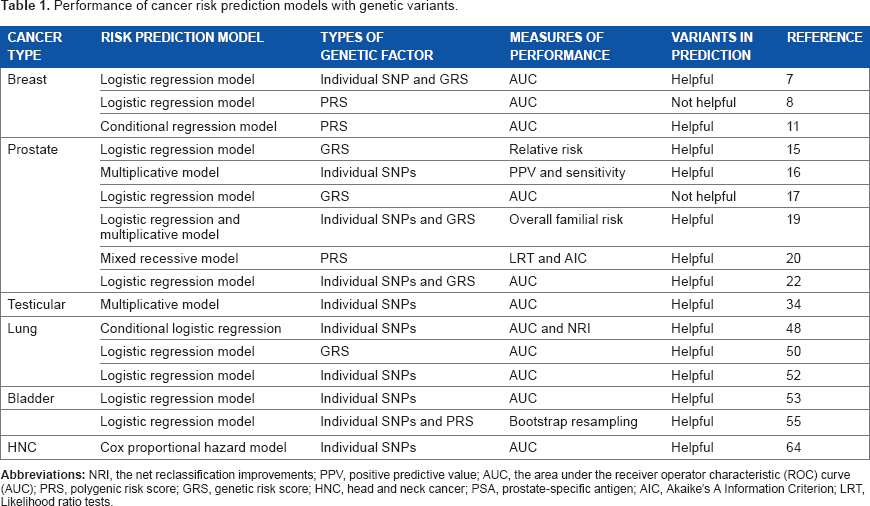
Table 2 summarizes the frequently used risk prediction models with genetic factors and general modeling procedures. The most commonly used model is logistic regression model. When dealing with multiple genetic factors and other covariates, logistic regression assumes a linear relationship among the predictors and uses a logit link to combine them into a one-dimensional fitted value.
The frequently used risk prediction models and general modeling procedures.

Although more than 50 cancer GWAS incorporating more than 15 different malignancies have been reported, identifying over 100 genomic cancer susceptibility regions,
3
for most malignancies the number of consistently confirmed SNPs is less than a dozen. The lack of power of GWAS suggests that there may exist many more SNPs associated with some malignancies that have smaller effect sizes. However, such SNPs may be statistically insignificant in genome-wide. How to effectively incorporate these SNPs in a risk prediction model is challenging since a group of these SNPs may likely make a positive contribution to a risk prediction model while the other ones may just add some noise. Evans et al.
18
and Purcell et al.
65
have proposed methods of aggregating information on a large number of SNP alleles associated with a trait that does not achieve stringent genome-wide statistical significance or even nominal statistical significance of
Compared with traditional risk factors such as family history, smoking, age, and sex, sometimes the impact of genetic variants in predicting risk is small which may reflect the small effect size of disease-associated SNPs integrated in the risk prediction model. Due to the difference in effect sizes of associated SNPs, the power of genetic variants in prediction for different cancers is different as well. A recent report uses PRS to estimate the relative risks of disease. In these reported estimates, the predictive power is higher for prostate cancer than for breast cancer, which reflects the fact that the known associated SNP effect sizes for prostate cancer are greater and account for a larger percentage of the familial relative risk. 66
In the article, we reviewed the cancer risk prediction models by different cancer types. But many cancers share the same major oncogenic or tumor suppressor genes such as
Author Contributions
Conceived and designed the experiments: XW, XH. Analyzed the data: XH, XZ. Wrote the first draft of the manuscript: XW, XH. Contributed to the writing of the manuscript: MO, XZ, DQ Agree with manuscript results and conclusions: XW, MO, XZ, XH, DQ Jointly developed the structure and arguments for the paper: XW, MO. Made critical revisions and approved final version: XW, MO. All authors reviewed and approved of the final manuscript.